Modulation of the Endocannabinoid System Following Central Nervous System Injury

 ,
, 
{kind=link}
{kind=link}
Abstract
1. Clinical Background and Pathophysiology
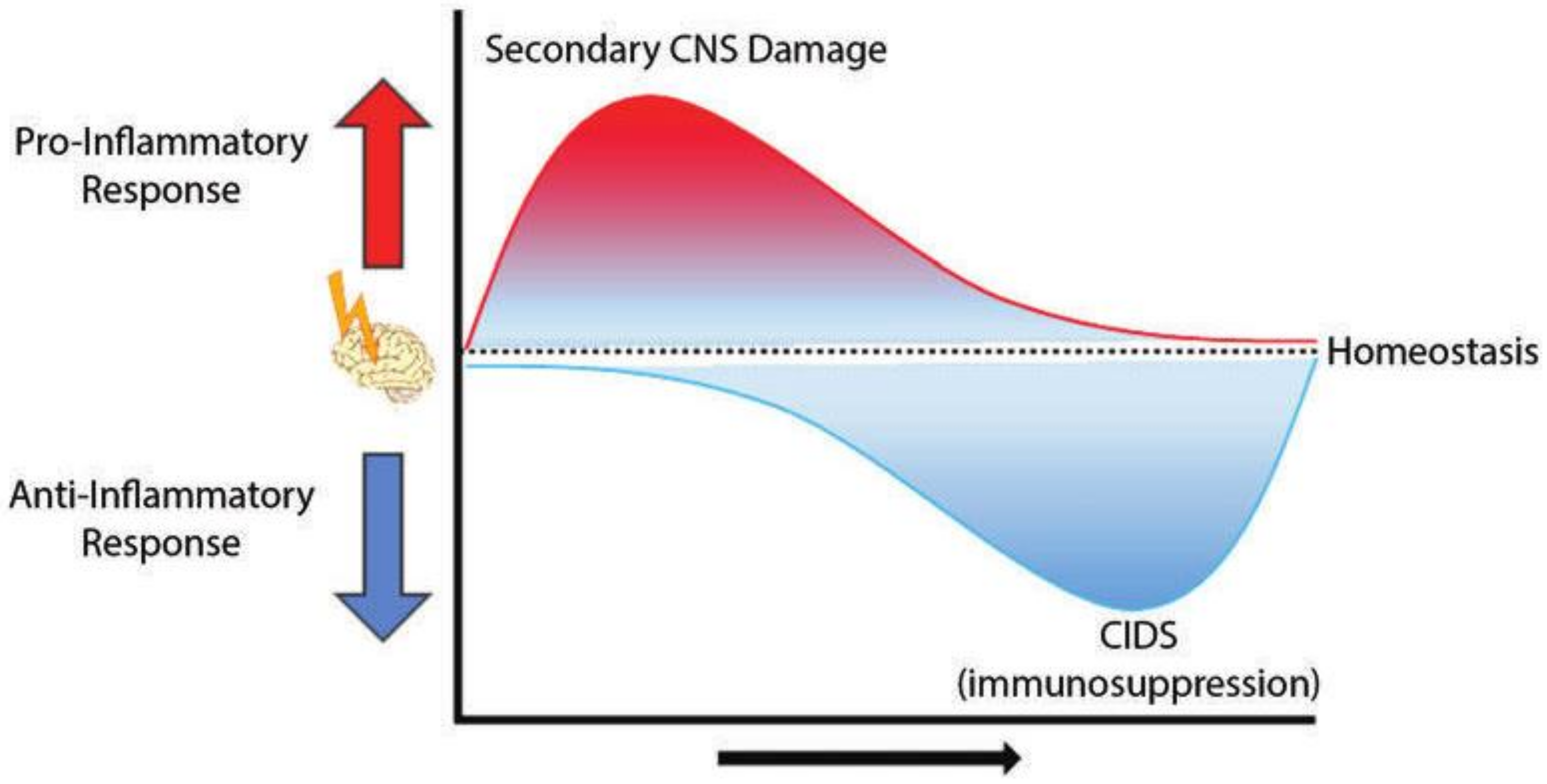
2. CNS Injury-Associated Immune Response
3. The Endocannabinoid System in CNS Injury
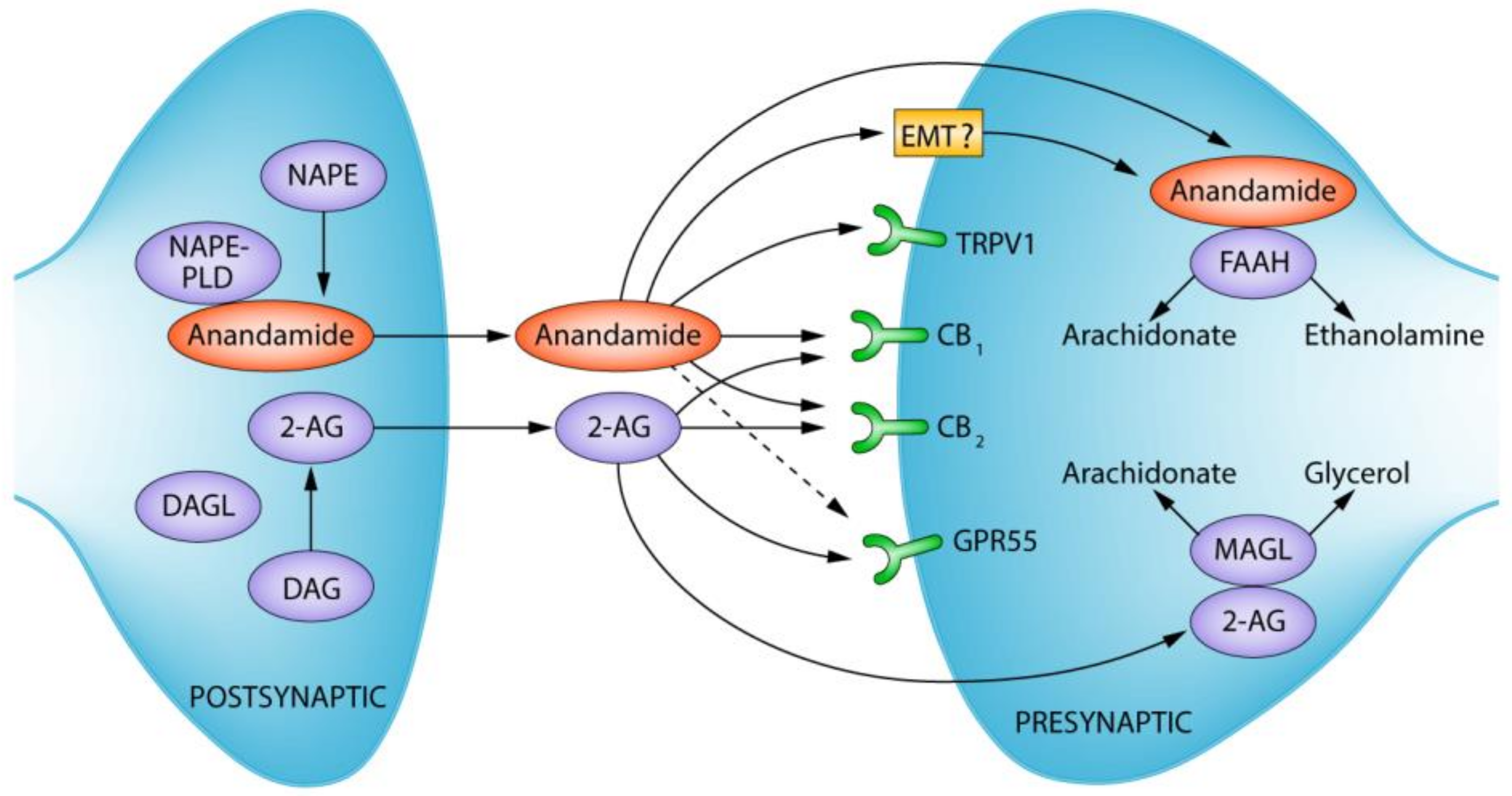
The Endocannabinoid System
4. Early Post-Stroke Phase
4.1. Overview of Pathophysiology
4.2. Potential for ECS Modulation
5. Late Post-Stroke Phase
5.1. Overview of Pathophysiology
5.2. Potential ECS Modulation During Late Post-Stroke Phase
6. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Thrift, A.G.; Thayabaranathan, T.; Howard, G.; Howard, V.J.; Rothwell, P.M.; Feigin, V.L.; Norrving, B.; Donnan, G.A.; Cadilhac, D.A. Global stroke statistics. Int. J. Stroke 2017, 12, 13–32. [Google Scholar] [CrossRef]
- Hankey, G. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; De Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics—2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dogra, S. Pathophysiology and therapeutic strategies in the management of stroke: An update. Drugs Today (Barc.) 2008, 44, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, A.; Valvassori, L. Endovascular treatment of acute ischemic stroke. N. Engl. J. Med. 2013, 368, 2433–2434. [Google Scholar] [CrossRef] [PubMed]
- Helbok, R.; Kurtz, P.; Schmidt, J.M.; Stuart, R.M.; Fernandez, L.; Malhotra, R.; Presciutti, M.; Ostapkovich, N.D.; Connolly, E.S.; Lee, K.; et al. Effect of mannitol on brain metabolism and tissue oxygenation in severe haemorrhagic stroke. J. Neurol. Neurosurg. Psychiatry 2011, 82, 378–383. [Google Scholar] [CrossRef]
- Balami, J.S.; Buchan, A.M. Complications of intracerebral haemorrhage. Lancet Neurol. 2012, 11, 101–118. [Google Scholar] [CrossRef]
- Hemphill, J.C.; Greenberg, S.M.; Anderson, C.S.; Becker, K.; Bendok, B.R.; Cushman, M.; Fung, G.L.; Goldstein, J.N.; MacDonald, R.L.; Mitchell, P.H.; et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2015, 46, 2032–2060. [Google Scholar] [CrossRef]
- Viitanen, M.; Winblad, B.; Asplund, K. Autopsy-verified Causes of Death after Stroke. Acta Med. Scand. 1987, 222, 401–408. [Google Scholar] [CrossRef]
- Langhorne, P.; Stott, J.D.; Robertson, L.; MacDonald, J.; Jones, L.; McAlpine, C.; Dick, F.; Taylor, S.G.; Murray, G. Medical Complications After Stroke. Stroke J. Am. Heart Assoc. 2000, 31, 1223–1229. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2012, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, A.; Becker, K.; Dressel, A. Immunological consequences of ischemic stroke. Acta Neurol. Scand. 2014, 129, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, Y. The yin and yang of innate immunity in stroke. BioMed Res. Int. 2014, 2014, 807978. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Petrovic-Djergovic, D.; Goonewardena, S.; Pinsky, D. Inflammatory disequilibrium in stroke. Circ. Res. 2016, 119, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Malone, K.; Amu, S.; Moore, A.C.; Waeber, C. The immune system and stroke: From current targets to future therapy. Immunol. Cell Biol. 2018. [Google Scholar] [CrossRef]
- Meisel, C.; Schwab, J.M.; Prass, K.; Meisel, A.; Dirnagl, U. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 2005, 6, 775–786. [Google Scholar] [CrossRef]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef]
- Klehmet, J.; Harms, H.; Richter, M.; Prass, K.; Volk, H.D.; Dirnagl, U.; Meisel, A.; Meisel, C. Stroke-induced immunodepression and post-stroke infections: Lessons from the preventive antibacterial therapy in stroke trial. Neuroscience 2009, 158, 1184–1193. [Google Scholar] [CrossRef]
- Shim, R.; Wong, C.H.Y. Ischemia, immunosuppression and infection-tackling the predicaments of post-stroke complications. Int. J. Mol. Sci. 2016, 17, 64. [Google Scholar] [CrossRef]
- Van de Beek, D.; Wijdicks, E.F.M.; Vermeij, F.H.; de Haan, R.J.; Prins, J.M.; Spanjaard, L.; Dippel, D.W.J.; Nederkoorn, P.J. Preventive Antibiotics for Infections in Acute Stroke. Arch. Neurol. 2009, 66, 1076–1081. [Google Scholar] [CrossRef]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside Antoine. Ther. Adv. Neurol. Disord. Immunol. 2018, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, Z.Y.; Huang, T.T.; Zhou, Y.X.; Wang, X.; Yang, L.Q.; Chen, Z.A.; Yu, W.F.; Li, P.Y. The peripheral immune response after stroke—A double edge sword for blood-brain barrier integrity. CNS Neurosci. Ther. 2018, 24, 1115–1128. [Google Scholar] [CrossRef]
- Kelly, M.E.; Lehmann, C.; Zhou, J. Immune modulation by cannabinoids during central nervous system injury-induced neuroinflammation. In The Endocannabinoid System in Local and Systemic Inflammation; Morgan & Claypool Publishers: Williston, VT, USA, 2017; pp. 97–107. [Google Scholar]
- Chu, H.X.; Kim, H.A.; Lee, S.; Moore, J.P.; Chan, C.T.; Vinh, A.; Gelderblom, M.; Arumugam, T.V.; Broughton, B.R.S.; Drummond, G.R.; et al. Immune cell infiltration in malignant middle cerebral artery infarction: Comparison with transient cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T Lymphocytes and Interferon-γ in Ischemic Stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.V.; Benakis, C.; Liesz, A. T cells in the post-ischemic brain: Troopers or paramedics? J. Neuroimmunol. 2019, 326, 33–37. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Kraft, P.; Dreykluft, A.; Hagedorn, I.; Gobel, K.; Schuhmann, M.K.; Langhauser, F.; Helluy, X.; Schwarz, T.; Bittner, S.; et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood 2013, 121, 679–691. [Google Scholar] [CrossRef]
- Ortega, S.; Noorbhai, I.; Poinsatte, K.; Kong, X.; Anderson, A.; Monson, N.; Stowe, A. Stroke Induces a Rapid Adaptive Autoimmune Response to Novel Neuronal Antigens. Discov. Med. 2015, 19, 381–392. [Google Scholar]
- Planas, A.M.; Go´mez-Choco, M.; Xabier, U.; Gorina, R.; Caballero, M.; Chamorro, A. Ngel Brain-Derived Antigens in Lymphoid Tissue of Patients with Acute Stroke Anna. J. Immunol. 2012, 188, 2156–2163. [Google Scholar] [CrossRef]
- Dirnagl, U.; Klehmet, J.; Braun, J.S.; Harms, H.; Meisel, C.; Ziemssen, T.; Prass, K.; Meisel, A. Stroke-induced immunodepression: Experimental evidence and clinical relevance. Stroke 2007, 38, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Pertwee, R.G. Endocannabinoids and Their Pharmacological Actions. Handb. Exp. Pharmacol. 2015, 231, 1–37. [Google Scholar] [PubMed]
- Matsuda, L.; Lolait, S.; Brownstein, M.; Young, A.; Bonner, T. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Lesley, A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Pertwee, R.G.; et al. Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease—Successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Steffens, S.; Haskó, G.; Schindler, T.H.; Kunos, G. Cardiovascular effects of marijuana and synthetic cannabinoids: The good, the bad, and the ugly. Nat. Rev. Cardiol. 2018, 15, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 761, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.-R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S. Commentary: Functional Neuronal CB2 Cannabinoid Receptors in the CNS. Curr. Neuropharmacol. 2011, 9, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; LE Fur, G.; Casellas, P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B. Critical Enzymes Involved in Endocannabinoid Metabolism. Protein Pept. Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Schicho, R.; Storr, M. Patients with IBD find symptom relief in the Cannabis field. Nat. Rev. Gastroenterol. Hepatol. 2013, 11, 142–143. [Google Scholar] [CrossRef] [PubMed]
- Lafreniere, J.D.; Kelly, M.E.M. Potential for Endocannabinoid System Modulation in Ocular Pain and Inflammation: Filling the Gaps in Current Pharmacological Options. Neuronal Signal. 2018, NS20170144. [Google Scholar] [CrossRef]
- Greineisen, W.E.; Turner, H. Immunoactive effects of cannabinoids: Considerations for the therapeutic use of cannabinoid receptor agonists and antagonists. Int. Immunopharmacol. 2010, 10, 547–555. [Google Scholar] [CrossRef]
- Toguri, J.T.; Lehmann, C.; Laprairie, R.B.; Szczesniak, A.M.; Zhou, J.; Denovan-Wright, E.M.; Kelly, M.E.M. Anti-inflammatory effects of cannabinoid CB(2) receptor activation in endotoxin-induced uveitis. Br. J. Pharmacol. 2014, 171, 1448–1461. [Google Scholar] [CrossRef]
- Croxford, J.L.; Miller, S.D. Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R+WIN55,212. J. Clin. Investig. 2003, 111, 1231–1240. [Google Scholar] [CrossRef]
- Yu, S.J.; Reiner, D.; Shen, H.; Wu, K.J.; Liu, Q.R.; Wang, Y. Time-dependent protection of CB2 receptor agonist in stroke. PLoS ONE 2015, 10, e0132487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Martin, B.R.; Adler, M.W.; Razdan, R.K.; Ganea, D.; Tuma, R.F. Modulation of the balance between cannabinoid CB1 and CB2 receptor activation during cerebral ischemic/reperfusion injury. Neuroscience 2008, 152, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The Endocannabinoid System and Oligodendrocytes in Health and Disease. Front. Neurosci. 2018, 12, 733. [Google Scholar] [CrossRef]
- Hillard, C.J. NIH Public Access. Curr. Pharm. Des. 2009, 14, 2347–2361. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Fernández-López, D.; García-Yébenes, I.; García-Gutiérrez, M.S.; Vivancos, J.; Nombela, F.; Torres, M.; Burguete, M.C.; Manzanares, J.; Lizasoain, I.; et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 2012, 43, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.A.; Raborn, E.S.; Griffin, L.; Dennis, J.; Marciano-Cabral, F. CB 2 receptors in the brain: Role in central immune function. Br. J. Pharmacol. 2008, 153, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Capettini, L.S.A.; Savergnini, S.Q.; Da Silva, R.F.; Stergiopulos, N.; Santos, R.A.S.; MacH, F.; Montecucco, F. Update on the role of cannabinoid receptors after ischemic stroke. Mediat. Inflamm. 2012, 2012, 824093. [Google Scholar] [CrossRef] [PubMed]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef]
- Zhang, M.; Martin, B.R.; Adler, M.W.; Razdan, R.K.; Jallo, J.I.; Tuma, R.F. Cannabinoid CB(2) receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J. Cereb. Blood Flow Metab. 2007, 27, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Adler, M.W.; Abood, M.E.; Ganea, D.; Jallo, J.; Tuma, R.F. CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc. Res. 2009, 78, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Ortega, Z.; Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. CB2 Cannabinoid Receptors Promote Neural Progenitor Cell Proliferation via mTORC1 Signaling. J. Biol. Chem. 2012, 287, 1198–1209. [Google Scholar] [CrossRef]
- Elliott, M.B.; Tuma, R.F.; Amenta, P.S.; Barbe, M.F.; Jallo, J.I. Acute Effects of a Selective Cannabinoid-2 Receptor Agonist on Neuroinflammation in a Model of Traumatic Brain Injury. J. Neurotrauma 2011, 28, 973–981. [Google Scholar] [CrossRef]
- Adhikary, S.; Li, H.; Heller, J.; Skarica, M.; Zhang, M.; Ganea, D.; Tuma, R.F. Modulation of inflammatory responses by a cannabinoid-2-selective agonist after spinal cord injury. J. Neurotrauma 2011, 28, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Dhopeshwarkar, A.; Mackie, K. CB2 Cannabinoid Receptors as a Therapeutic Target-What Does the Future Hold? Mol. Pharmacol. Mol. Pharmacol. 2014, 86, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Allan, S.M.; Rothwell, N.J. Inflammation in central nervous system injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1669–1677. [Google Scholar] [CrossRef]
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Hooper, D.C.; Elliott, M.B. Cannabinoid receptor type-2 stimulation, blockade, and deletion alter the vascular inflammatory responses to traumatic brain injury. J. Neuroinflamm. 2014, 11, 191–201. [Google Scholar] [CrossRef]
- England, T.J.; Hind, W.H.; Rasid, N.A.; O’sullivan, S.E. Cannabinoids in experimental stroke: A systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 2015, 35, 348–358. [Google Scholar] [CrossRef]
- Mishima, K.; Hayakawa, K.; Abe, K.; Ikeda, T.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism. Stroke 2005, 36, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martínez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT1A and CB2 receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Hind, W.H.; England, T.J.; O’Sullivan, S.E. Cannabidiol protects an in vitro model of the blood-brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT 1A receptors. Br. J. Pharmacol. 2016, 173, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Tham, M.; Yilmaz, O.; Alaverdashvili, M.; Kelly, M.E.M.; Denovan-Wright, E.M.; Laprairie, R.B. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Parmentier-Batteur, S.; Jin, K.; Mao, X.O.; Xie, L.; Greenberg, D. A Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J. Neurosci. 2002, 22, 9771–9775. [Google Scholar] [CrossRef]
- Caltana, L.; Saez, T.M.; Aronne, M.P.; Brusco, A. Cannabinoid receptor type 1 agonist ACEA improves motor recovery and protects neurons in ischemic stroke in mice. J. Neurochem. 2015, 135, 616–629. [Google Scholar] [CrossRef]
- Muthian, S.; Rademacher, D.J.; Roelke, C.T.; Gross, G.J.; Hillard, C.J. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience 2004, 129, 743–750. [Google Scholar] [CrossRef]
- Ward, S.J.; Castelli, F.; Reichenbach, Z.W.; Tuma, R.F. Surprising outcomes in cannabinoid CB1/CB2 receptor double knockout mice in two models of ischemia. Life Sci. 2018, 195, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.S.V.; Kelly, M.E.M. Cannabinoids in the Cardiovascular System, 1st ed.; Elsevier Inc.: New York, NY, USA, 2017; Volume 80, ISBN 9780128112328. [Google Scholar]
- Kamel, H.; Iadecola, C. Brain-immune interactions and ischemic stroke: Clinical implications. Arch. Neurol. 2012, 69, 576–581. [Google Scholar]
- Liu, D.-D.; Chu, S.-F.; Chen, C.; Yang, P.-F.; Chen, N.-H.; He, X. Research progress in stroke-induced immunodepression syndrome (SIDS) and stroke-associated pneumonia (SAP). Neurochem. Int. 2018, 114, 42–54. [Google Scholar] [CrossRef]
- Wang, X. Investigational anti-inflammatory agents for the treatment of ischaemic brain injury Investigational anti-inflammatory agents for the treatment of ischaemic brain injury. Expert Opin. Investig. Drugs 2005, 14, 393–409. [Google Scholar] [CrossRef]
- Burkovskiy, I.; Zhou, J.; Lehmann, C. Experimental Cannabinoid 2 Receptor Inhibition in CNS Injury-Induced Immunodeficiency Syndrome. Microcirculation 2016, 23, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Orgado, J.M.; Fernández-Ruiz, J.; Romero, J. The endocannabinoid system in neuropathological states. Int. Rev. Psychiatry 2009, 21, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Rivers-Auty, J.R.; Smith, P.F.; Ashton, J.C. The cannabinoid CB2receptor agonist GW405833 does not ameliorate brain damage induced by hypoxia-ischemia in rats. Neurosci. Lett. 2014, 569, 104–109. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Noori, H.; Burkovskiy, I.; Lafreniere, J.D.; Kelly, M.E.M.; Lehmann, C. Modulation of the Endocannabinoid System Following Central Nervous System Injury. Int. J. Mol. Sci. 2019, 20, 388. https://doi.org/10.3390/ijms20020388
Zhou J, Noori H, Burkovskiy I, Lafreniere JD, Kelly MEM, Lehmann C. Modulation of the Endocannabinoid System Following Central Nervous System Injury. International Journal of Molecular Sciences. 2019; 20(2):388. https://doi.org/10.3390/ijms20020388
Chicago/Turabian StyleZhou, Juan, Haneen Noori, Ian Burkovskiy, J. Daniel Lafreniere, Melanie E. M. Kelly, and Christian Lehmann. 2019. "Modulation of the Endocannabinoid System Following Central Nervous System Injury" International Journal of Molecular Sciences 20, no. 2: 388. https://doi.org/10.3390/ijms20020388
APA StyleZhou, J., Noori, H., Burkovskiy, I., Lafreniere, J. D., Kelly, M. E. M., & Lehmann, C. (2019). Modulation of the Endocannabinoid System Following Central Nervous System Injury. International Journal of Molecular Sciences, 20(2), 388. https://doi.org/10.3390/ijms20020388