Epitope Mapping Immunoassay Analysis of the Interaction between β-Amyloid and Fibrinogen



Abstract
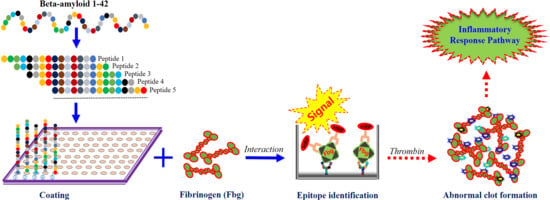
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Preparation of Fbg and Aβ42 EpiMap ELISA Tests
2.2. The Evaluation of Specificity and Sensitivity of the Antibodies
2.3. Protein Epitope Mapping with the EpiMap ELISA
2.4. Fbg Binding Analysis Using Aβ42 Epitope-Mapping ELISA
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. The Preparation of the Aβ42 Fragments and Aβ42 EpiMap ELISA Tests
4.3. Fbg and Its Fragments Binding Assay Using the Anti-Fbg Antibody
4.4. ELISA Aβ42–Fbg Interaction Assay
4.5. SDS-Stable Complex Formation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S. Clinical genetic strategies for early onset neurodegenerative diseases. Mol. Cell. Toxicol. 2018, 14, 123–142. [Google Scholar] [CrossRef]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.Y.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Yi, S.; Han, J.Y.; Park, S.Y.; Jang, J.W.; Chun, I.K.; Giau, V.V.; Bagyinszky, E.; Lim, K.T.; Kang, S.M.; et al. Analysis of Cerebrospinal Fluid and [11C]PIB PET Biomarkers for Alzheimer’s Disease with Updated Protocols. J. Alzheimer’s Dis. 2016, 52, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299. [Google Scholar] [PubMed]
- Zamolodchikov, D.; Strickland, S. Abeta delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 2012, 119, 3342–3351. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Giau, V.V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Mattei, L.; Richards, A.T.; Norris, E.H.; Strickland, S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol. Aging 2015, 36, 608–617. [Google Scholar] [CrossRef]
- Hultman, K.; Cortes-Canteli, M.; Bounoutas, A.; Richards, A.T.; Strickland, S.; Norris, E.H. Plasmin deficiency leads to fibrin accumulation and a compromised inflammatory response in the mouse brain. J. Thromb. Haemost. 2014, 12, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Klohs, J.; Baltes, C.; Princz-Kranz, F.; Ratering, D.; Nitsch, R.M.; Knuesel, I.; Rudin, M. Contrast-enhanced magnetic resonance microangiography reveals remodeling of the cerebral microvasculature in transgenic ArcAbeta mice. J. Neurosci. 2012, 32, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between beta-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. [Google Scholar] [CrossRef]
- Felding-Habermann, B.; Ruggeri, Z.M.; Cheresh, D.A. Distinct biological consequences of integrin alpha v beta 3-mediated melanoma cell adhesion to fibrinogen and its plasmic fragments. J. Biol. Chem. 1992, 267, 5070–5077. [Google Scholar] [PubMed]
- Lichtenthaler, S.F. alpha-secretase in Alzheimer’s disease: Molecular identity, regulation and therapeutic potential. J. Neurochem. 2011, 116, 10–21. [Google Scholar] [CrossRef]
- Manea, M.; Przybylski, M.; Hudecz, F.; Mezo, G. Design, structural, and immuno-analytical properties of antigenic bioconjugates comprising a beta-amyloid-plaque specific epitope. Biopolymers 2008, 90, 94–104. [Google Scholar] [CrossRef]
- Stefanescu, R.; Lupu, L.; Manea, M.; Iacob, R.E.; Przybylski, M. Molecular characterization of the beta-amyloid(4-10) epitope of plaque specific Abeta antibodies by affinity-mass spectrometry using alanine site mutation. J. Peptide Sci. 2018, 24, e3047. [Google Scholar] [CrossRef]
- Barghorn, S.; Nimmrich, V.; Striebinger, A.; Krantz, C.; Keller, P.; Janson, B.; Bahr, M.; Schmidt, M.; Bitner, R.S.; Harlan, J.; et al. Globular amyloid beta-peptide oligomer—A homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 2005, 95, 834–847. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Holmbäck, K.; Danton, M.J.; Suh, T.T.; Daugherty, C.C.; Degen, J.L. Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin alpha IIb beta 3. EMBO J. 1996, 15, 5760–5771. [Google Scholar] [CrossRef] [PubMed]
- Oki, T.; Kitaura, J.; Eto, K.; Lu, Y.; Maeda-Yamamoto, M.; Inagaki, N.; Nagai, H.; Yamanishi, Y.; Nakajima, H.; Kumagai, H.; et al. Integrin alphaIIbbeta3 induces the adhesion and activation of mast cells through interaction with fibrinogen. J. Immunol. 2006, 176, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Lishko, V.K.; Podolnikova, N.P.; Yakubenko, V.P.; Yakovlev, S.; Medved, L.; Yadav, S.P.; Ugarova, T.P. Multiple binding sites in fibrinogen for integrin alphaMbeta2 (Mac-1). J. Biol. Chem. 2004, 279, 44897–44906. [Google Scholar] [CrossRef] [PubMed]
- Nham, S.U. Characteristics of fibrinogen binding to the domain of CD11c, an alpha subunit of p150,95. Biochem. Biophys. Res. Commun. 1999, 264, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Laudano, A.P.; Doolittle, R.F. Studies on synthetic peptides that bind to fibrinogen and prevent fibrin polymerization. Structural requirements, number of binding sites, and species differences. Biochemistry 1980, 19, 1013–1019. [Google Scholar] [CrossRef]
- Munson, G.W.; Roher, A.E.; Kuo, Y.M.; Gilligan, S.M.; Reardon, C.A.; Getz, G.S.; LaDu, M.J. SDS-stable complex formation between native apolipoprotein E3 and beta-amyloid peptides. Biochemistry 2000, 39, 16119–16124. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef]
- Youn, Y.C.; Lim, Y.K.; Han, S.H.; Giau, V.V.; Lee, M.K.; Park, K.Y.; Kim, S.; Bagyinszky, E.; An, S.S.A.; Kim, H.R. Apolipoprotein epsilon7 allele in memory complaints: Insights through protein structure prediction. Clin. Interv. Aging 2017, 12, 1095–1102. [Google Scholar] [CrossRef]
- Ahn, H.J.; Glickman, J.F.; Poon, K.L.; Zamolodchikov, D.; Jno-Charles, O.C.; Norris, E.H.; Strickland, S. A novel Abeta-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J. Exp. Med. 2014, 211, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Namkoong, H.; Kim, H.K.; Kim, S.; Hwang, D.W.; Na, H.R.; Ha, S.-A.; Kim, J.-R.; Kim, J.W. Fibrinogen gamma-A chain precursor in CSF: A candidate biomarker for Alzheimer’s disease. BMC Neurol. 2007, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, H.; Zhang, S.; Fan, X.; Liu, X. Plasma fibrinogen is associated with cognitive decline and risk for dementia in patients with mild cognitive impairment. Int. J. Clin. Pract. 2008, 62, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Muradashvili, N.; Tyagi, R.; Metreveli, N.; Tyagi, S.C.; Lominadze, D. Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen-amyloid beta complex formation during hyperhomocysteinemia. J. Cereb. Blood Flow Metab. 2014, 34, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Giau, V.V.; Vo, T.K. Current advances in transdermal delivery of drugs for Alzheimer’s disease. Indian J. Pharmacol. 2017, 49, 145–154. [Google Scholar] [PubMed]
- Kang, M.; Kim, S.Y.; An, S.S.; Ju, Y.R. Characterizing affinity epitopes between prion protein and beta-amyloid using an epitope mapping immunoassay. Exp. Mol. Med. 2013, 45, e34. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Giau, V.; An, S.S.A. Epitope Mapping Immunoassay Analysis of the Interaction between β-Amyloid and Fibrinogen. Int. J. Mol. Sci. 2019, 20, 496. https://doi.org/10.3390/ijms20030496
Van Giau V, An SSA. Epitope Mapping Immunoassay Analysis of the Interaction between β-Amyloid and Fibrinogen. International Journal of Molecular Sciences. 2019; 20(3):496. https://doi.org/10.3390/ijms20030496
Chicago/Turabian StyleVan Giau, Vo, and Seong Soo A. An. 2019. "Epitope Mapping Immunoassay Analysis of the Interaction between β-Amyloid and Fibrinogen" International Journal of Molecular Sciences 20, no. 3: 496. https://doi.org/10.3390/ijms20030496
APA StyleVan Giau, V., & An, S. S. A. (2019). Epitope Mapping Immunoassay Analysis of the Interaction between β-Amyloid and Fibrinogen. International Journal of Molecular Sciences, 20(3), 496. https://doi.org/10.3390/ijms20030496