Quantitative Proteomic Analysis of Castor (Ricinus communis L.) Seeds During Early Imbibition Provided Novel Insights into Cold Stress Response

Abstract
1. Introduction
2. Results
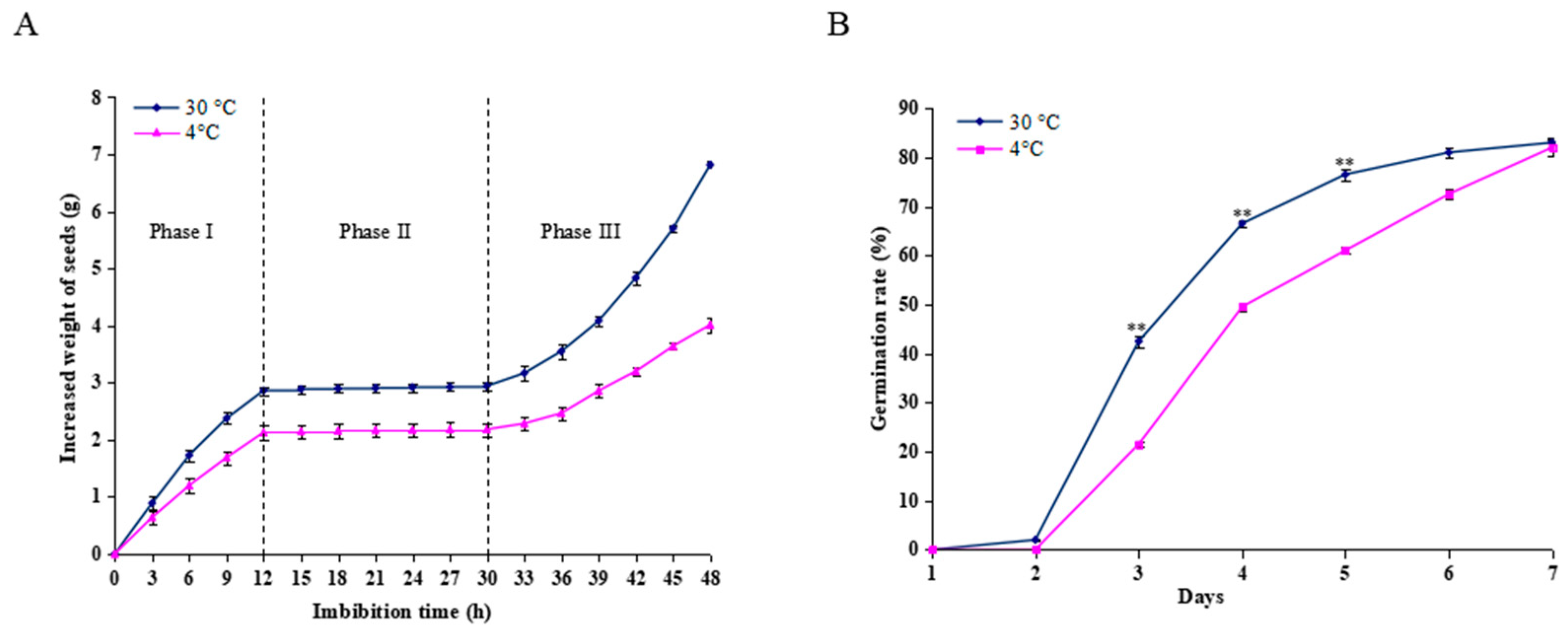
2.1. Germination Analysis of Castor Beans During Imbibition
2.2. Primary Data Analysis and Protein Identification
2.3. Identification of DAPS by iTRAQ
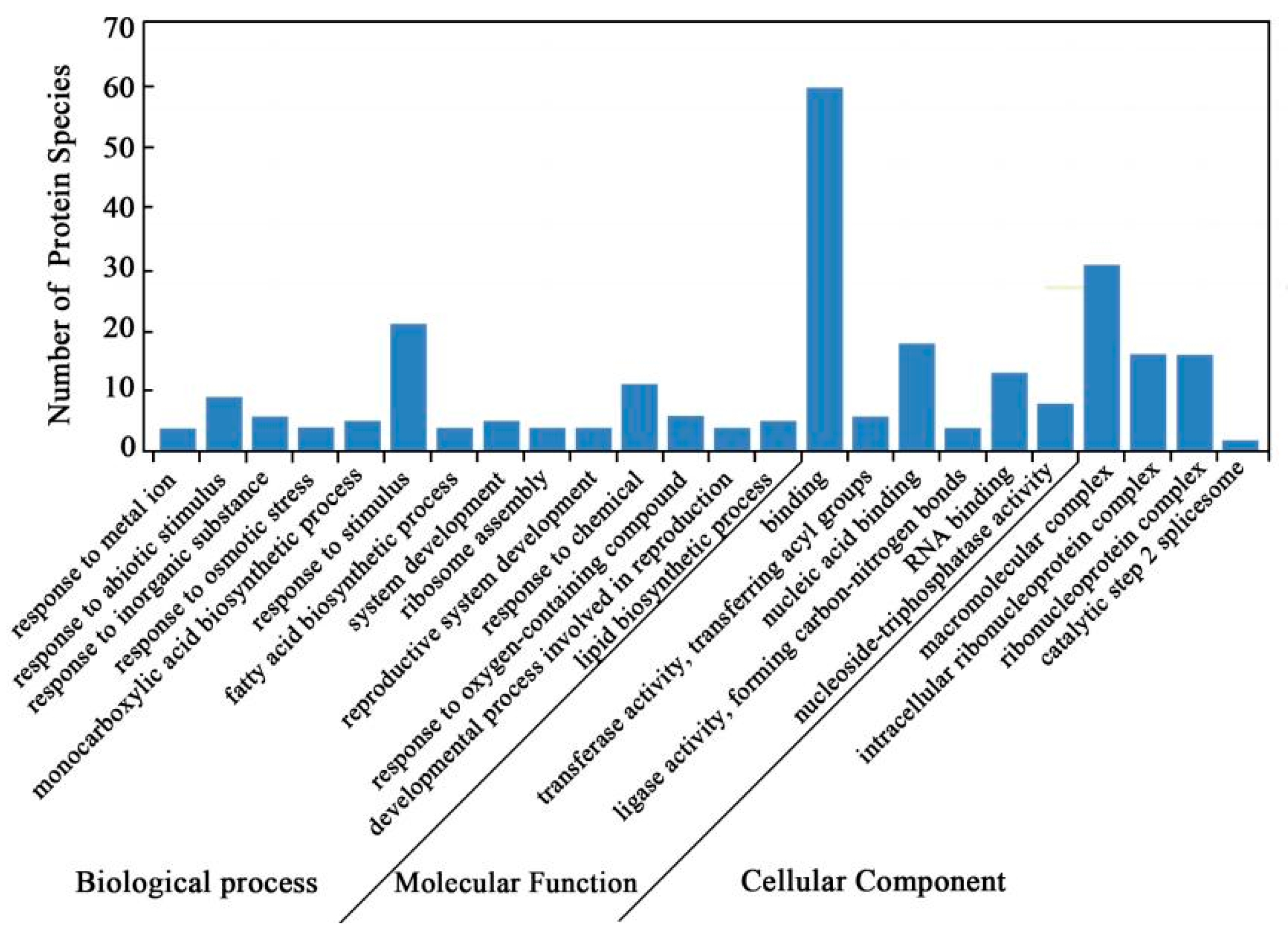
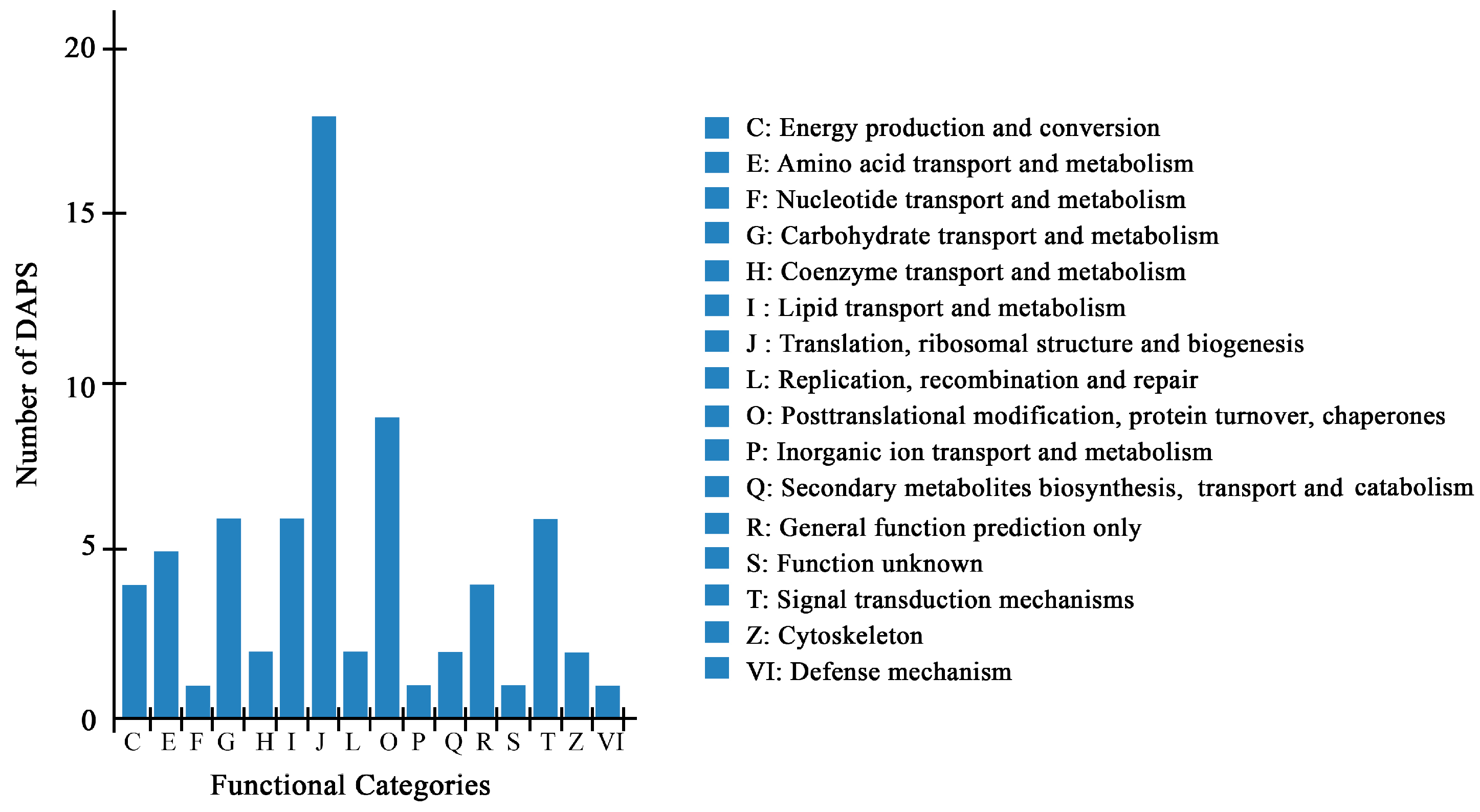
2.4. Bioinformatics Analysis of DAPS Identified by iTRAQ
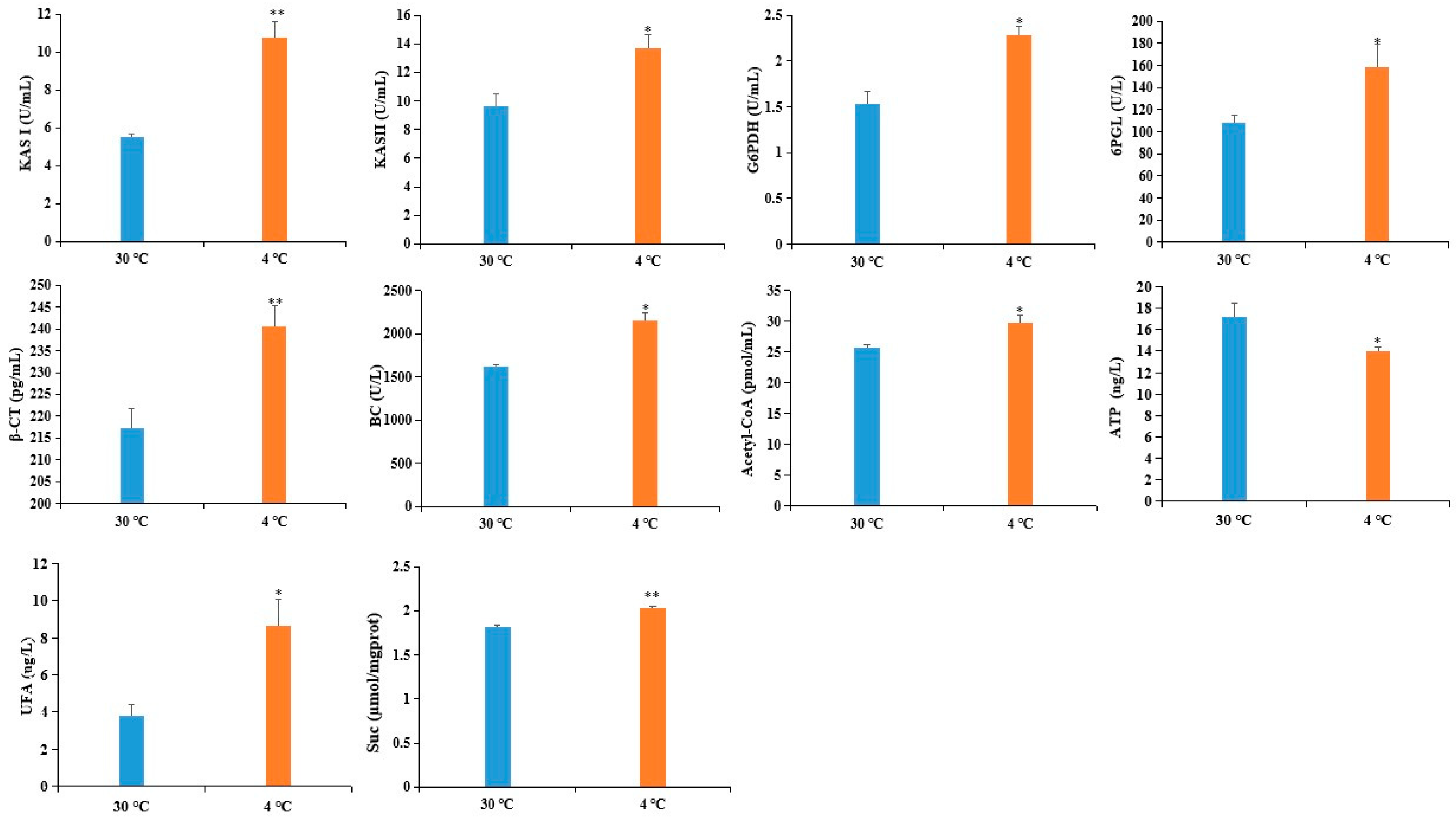
2.5. Confirmation of DAPS by ELISA
2.6. Transcriptional Analyses of the Corresponding Genes Encoding DAPS
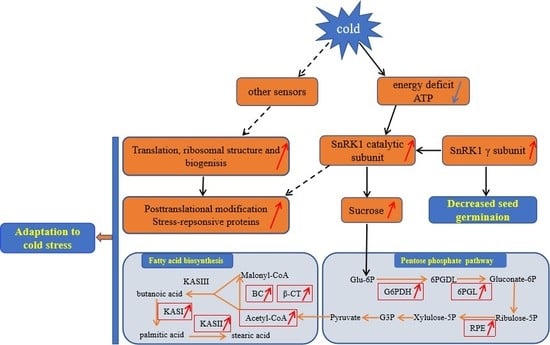
3. Discussion
3.1. DAPS Involved in Translation and Posttranslational Modification
3.2. DAPS Involved in Stress Response
3.3. DAPS Involved in Carbohydrate and Energy Metabolism
3.4. DAPS Involved in Lipid Transport and Metabolism
3.5. DAPS Involved in Signal Transduction
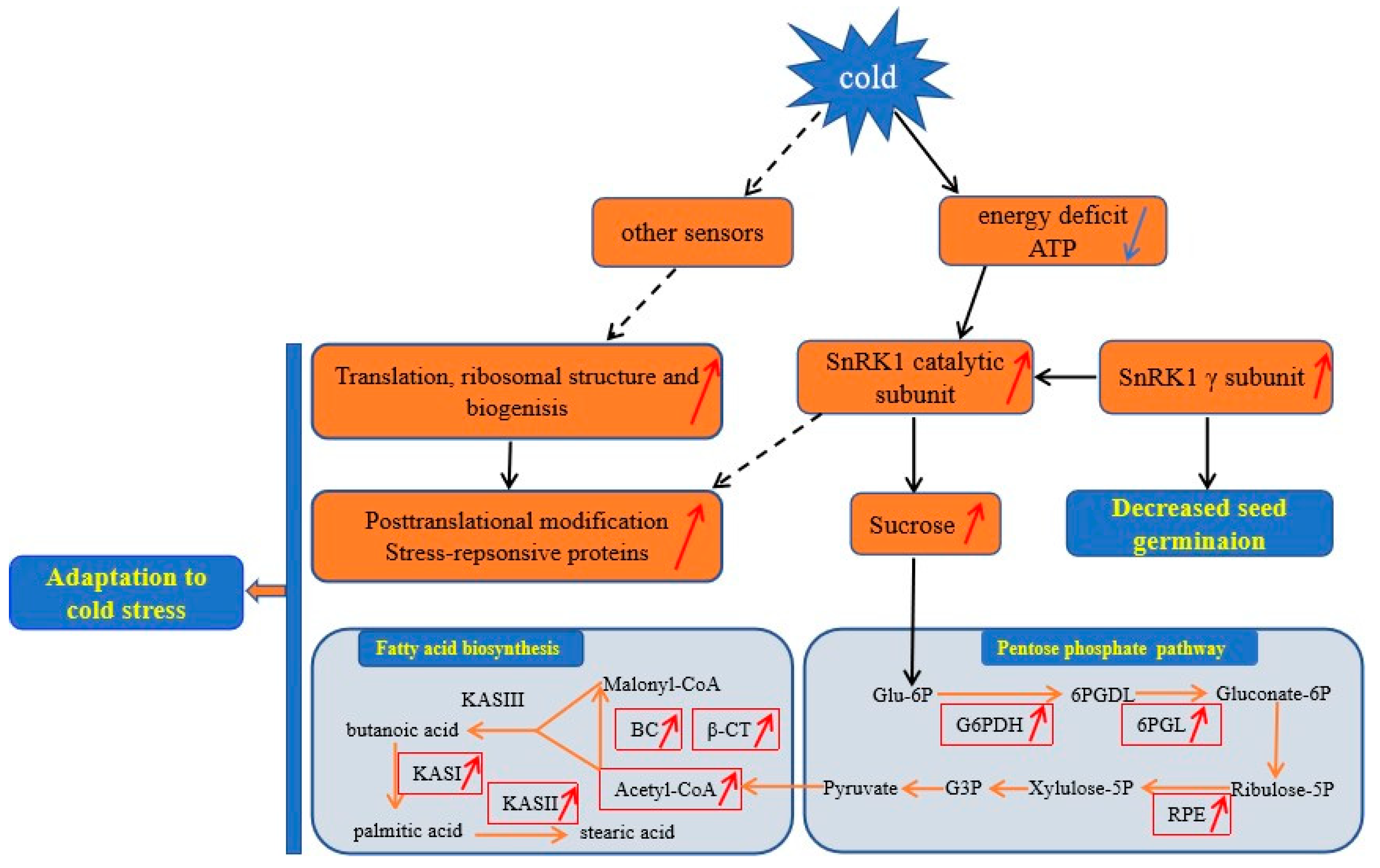
3.6. A Proposed Metabolic Pathway for Ricinus Communis During Early Seed Imbibition in Response to Cold Stress
4. Materials and Methods
4.1. Plant Materials and Stress Treatment
4.2. Protein Extraction
4.3. Protein Digestion and iTRAQ Labeling
4.5. Fractionation by SCX
4.6. LC-ESI-MS/MS Analysis by Q-Exactive
4.7. Protein Identification and Quantification
4.8. Bioinformatics Analysis
4.9. Enzyme-Linked Immunosorbent Assays
4.10. RNA Extraction and qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| iTRAQ | Isobaric tag for relative and absolute quantification |
| DAPS | Differential abundance protein species |
| ELISA | Enzyme-linked immunosorbent assays |
| DEGs | Differentially expressed genes |
| 2-DE | Two-dimensional electrophoresis |
| GO | Gene ontology |
| COG | Clusters of Orthologous Groups of proteins |
| KAS | β-ketoacyl-acyl carrier protein synthase |
| 6PGL | 6-phosphogluconolactonase |
| G6PDH | Glucose-6-phosphate 1-dehydrogenase |
| GPX | Glutathione peroxidase |
| LEA | Late embryogenesis abundant protein |
| SnRK1 | SNF1-related protein kniase |
| IF5A | Eukaryotic translation initiation factor 5A |
| PP2C | Protein phosphatase 2c |
References
- Wang, X.Y.; Shan, X.H.; Wu, Y.; Su, S.Z.; Li, S.P.; Liu, H.K.; Han, J.Y.; Xue, C.M.; Yuan, Y.P. iTRAQ-based quantitative proteomic analysis reveals new metabolic pathways responding to chilling stress in maize leaves. J. Proteom. 2016, 146, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.H.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef]
- Scholz, V.; da Silva, J.N. Prospects and risks of the use of castor oil as a fuel. Biomass Bioenergy 2008, 32, 95–100. [Google Scholar] [CrossRef]
- Lima Da Silva, N.; Maciel, M.; Batistella, C.; Filho, R. Optimization of biodiesel production from castor oil. Appl. Biochem. Biotechnol. 2006, 130, 405–414. [Google Scholar] [CrossRef]
- Jiang, X.J.; Wen, X.D. The effect of temperature on the germination rates in castor bean. Seed 2008, 27, 67–69. [Google Scholar]
- Wang, Z.F.; Wang, J.F.; Bao, Y.M.; Wu, Y.Y.; Zhang, H.S. Quantitative trait loci controlling rice seed germination under salt stress. Euphytica 2011, 178, 297–307. [Google Scholar] [CrossRef]
- Dong, K.; Zhen, S.M.; Cheng, Z.W.; Cao, H.; Ge, P.; Yan, Y.M. Proteomic analysis reveals key proteins and phosphoproteins upon seed germination of wheat (Triticum maestivum L.). Front. Plant Sci. 2015, 6, 1017. [Google Scholar] [CrossRef] [PubMed]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef]
- Pradet-Balade, B.; Boulme, F.; Beug, H.; Mullner, E.W.; Garcia-Sanz, J.A. Translation control: Bridging the gap between genomics and proteomics? Trends Biochem. Sci. 2001, 26, 225–229. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef]
- Kosová, K.; Vítámvás, P.; Prášil, I.T.; Renaut, J. Plant proteome changes under abiotic stress—Contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Gao, X.; Li, S.; Shi, M.; Javeed, H.; Jing, X.M.; Yang, G.X.; He, G.Y. Proteomic analysis of soybean (Glycine max (L.) Meer.) seeds during imbibition at chilling temperature. Mol. Breed. 2010, 26, 1–17. [Google Scholar] [CrossRef]
- Kosmala, A.; Bocian, A.; Rapacz, M.; Jurczyk, B.; Zwierzykowski, Z. Identification of leaf proteins differentially accumulated during cold acclimation between Festuca pratensis plants with distinct levels of frost tolerance. J. Exp. Bot. 2009, 60, 3595–3609. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zeng, X.; Wu, J.; Zhang, F.Q.; Li, C.X.; Jiang, J.J.; Wang, Y.P.; Sun, W.C. iTRAQ-based quantitative proteome revealed metabolic changes in winter turnip rape (Brassica rapa L.) under cold stress. Int. J. Mol. Sci. 2018, 19, 3346. [Google Scholar] [CrossRef]
- Maltman, D.J.; Simon, W.J.; Wheeler, C.H.; Dunn, M.J.; Wait, R.; Slabas, A.R. Proteomic analysis of the endoplasmic reticulum from developing and germinating seed of castor (Ricinus communis). Electrophoresis 2002, 23, 626–639. [Google Scholar] [CrossRef]
- Maltman, D.J.; Gadd, S.M.; Simon, W.J.; Slabas, A.R. Differential proteomic analysis of the endoplasmic reticulum from developing and germinating seeds of castor (Ricinus communis) identifies seed protein precursors as significant components of the endoplasmic reticulum. Proteomics 2007, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Houston, N.L.; Hajduch, M.; Thelen, J.J. Quantitative proteomics of seed filling in castor: Comparison with soybean and rapeseed reveals differences between photosynthetic and nonphotosynthetic seed metabolism. Plant Physiol. 2009, 151, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.A.P.; Nogueira, F.C.S.; Cardoso, K.C.; Costa, G.C.L.; Del Bem, L.E.V.; Domont, G.B.; Da Silva, M.J.; Moreira, R.C.; Soares, A.A.; Juca, T.L. Proteome analysis of castor bean seeds. Pure Appl. Chem. 2010, 82, 259–267. [Google Scholar] [CrossRef]
- Nogueira, F.C.; Palmisano, G.; Soares, E.L.; Shah, M.; Soares, A.A.; Roepstorff, P.; Campos, F.A.; Domont, G.B. Proteomic profile of the nucellus of castor bean (Ricinus communis L.) seeds during development. J. Proteom. 2012, 75, 1933–1939. [Google Scholar] [CrossRef]
- Nogueira, F.C.S.; Palmisano, G.; Schwämmle, V.; Soares, E.L.; Soares, A.A.; Roepstorff, P.; Domont, G.B.; Campos, F.A.P. Isotope labeling-based quantitative proteomics of developing seeds of castor oil seed (Ricinus communis L.). J. Proteome Res. 2013, 12, 5012–5024. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, H.L.; Ning, L.Y.; Li, B.; Bao, M.Z. Quantitative proteomic analysis provides novel insights into cold stress response in petunia seedlings. Front. Plant Sci. 2016, 7, 136. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.J.; Yan, X.C.; Wang, L.; Tan, M.L.; Geng, X.X.; Wei, W.H. Transcriptome analysis of the germinated seeds identifies low-temperature responsive genes involved in germination process in Ricinus communis. Acta Physiol. Plant 2016, 38, 6. [Google Scholar] [CrossRef]
- Sano, N.; Permana, H.; Kumada, R.; Shinozaki, Y.; Tanabata, T.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Proteomic analysis of embryonic proteins synthesized from long-lived mRNAs during germination of rice seeds. Plant Cell Physiol. 2012, 53, 687–698. [Google Scholar] [CrossRef]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression changes of ribosomal proteins in phosphate and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genom. 2013, 14, 783. [Google Scholar] [CrossRef]
- Kim, K.Y.; Park, S.W.; Chung, Y.S.; Chung, C.H.; Kim, J.I.; Lee, J.H. Molecular cloning of low-temperature-inducible ribosomal proteins from soybean. J. Exp. Bot. 2004, 55, 1153–1155. [Google Scholar] [CrossRef]
- Bukovnik, U.; Fu, J.; Bennett, M.; Prasad, P.V.; Ristic, Z. Heat tolerance and expression of protein synthesis elongation factors, EF-Tu and EF-1α, in spring wheat. Funct. Plant Biol. 2009, 36, 234–241. [Google Scholar] [CrossRef]
- Cui, S.; Huang, F.; Wang, J.; Ma, X.; Cheng, Y.; Liu, Y.J. A proteomic analysis of cold stress responses in rice seedlings. Proteomics 2005, 5, 3162–3172. [Google Scholar] [CrossRef]
- Lee, D.G.; Ahsan, N.; Lee, S.H.; Kang, K.Y.; Bahk, J.D.; Lee, I.J.; Lee, B.H. A proteomic approach in analyzing heat-responsive proteins in rice leaves. Proteomics 2007, 7, 3369–3383. [Google Scholar] [CrossRef]
- Li, A.L.; Li, H.Y.; Jin, B.F.; Ye, Q.N.; Zhou, T.; Yu, X.D.; Pan, X.; Man, J.H.; He, K.; Yu, M.; et al. A novel eIF5A complex functions as a regulator of p53 and p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 49251–49258. [Google Scholar] [CrossRef]
- Hopkins, M.T.; Lampi, Y.; Wang, T.W.; Liu, Z.; Thompson, J.E. Eukaryotic translation initiation factor 5A is involved in pathogen-induced cell death and development of disease symptoms in Arabidopsis. Plant Physiol. 2008, 148, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Lu, L.; Zhang, C.G.; Taylor, C.; Thompson, J.E. Pleiotropic effects of suppressing deoxyhypusine synthase expression in Arabidopsis thaliana. Plant Mol. Biol. 2003, 52, 1223–1235. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, B.; Jiang, C.; Ming, F. RceIF5A, encoding an eukaryotic translation initiation factor 5A in Rosa chinensis, can enhance thermotolerance, oxidative and osmotic stress resistance of Arabidopsis thaliana. Plant Mol. Biol. 2011, 75, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Q.; Xu, C.X.; Wang, C.; Wang, Y.C. Characterization of a eukaryotic translation initiation factor 5A homolog from Tamarix androssowii involved in plant abiotic stress tolerance. BMC Plant Biol. 2012, 12, 118. [Google Scholar] [CrossRef]
- Anand, M.; Chakraburtty, K.; Marton, M.J.; Hinnebusch, A.G.; Kinzy, T.G. Functional interactions between yeast translation eukaryotic elongation factor (eEF)1A and eEF3. J. Biol. Chem. 2003, 278, 6985–6991. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Moon, S.J.; Park, S.R.; Kim, B.G.; Byun, M.O. Elongation factor 1α from A. thaliana functions as molecular chaperone and confers resistance to salt stress in yeast and plants. Plant Sci. 2009, 177, 156–160. [Google Scholar] [CrossRef]
- Jeong, H.J.; Shin, J.S.; Ok, S.H. Barley DNA-binding methionine aminopeptidase, which changes the localization from the nucleus to the cytoplasm by low temperature, is involved in freezing tolerance. Plant Sci. 2011, 180, 53–60. [Google Scholar] [CrossRef]
- Mazzucotellin, E.; Mastrangelo, A.M.; Crosatti, C.; Guerra, D.; Stanca, A.M.; Cattivelli, L. Abiotic stress response in plants: When post-transcriptional and post-translational regulations control transcription. Plant Sci. 2008, 174, 420–431. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Callis, J.; Vierstra, R.D. Protein degradation in signaling. Curr. Opin. Plant Biol. 2000, 3, 381–386. [Google Scholar] [CrossRef]
- Zhou, G.A.; Chang, R.Z.; Qiu, L.J. Overexpression of soybean ubiquitin-conjugating enzyme gene GmUBC2 confers enhanced drought and salt tolerance through modulating abiotic stress-responsive gene expression in Arabidopsis. Plant Mol. Biol. 2010, 72, 357–367. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Tzavelas, C.; Pemberton, A.J.; Nezis, I.P.; Rivett, A.J.; Gonos, E.S. Overexpression of proteasome 5 subunit increases the amount of assembled proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem. 2005, 280, 11840–11850. [Google Scholar] [CrossRef] [PubMed]
- Parsell, D.A.; Lindquist, S. The function of heat-shock proteins in stress tolerance: Degradation and reactivation of damaged proteins. Annu. Rev. Genet. 1993, 27, 437–496. [Google Scholar] [CrossRef]
- Wang, W.X.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, O.; Virolainen, E.; Fagerstedt, K. Antioxidants, oxidative damage and oxygen deprivation stress: A review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Milla, M.A.R.; Maurer, A.; Huete, A.R.; Gustafson, J.P. Glutathione peroxidase genes in Arabidopsis are ubiquitous and regulated by abiotic stresses through diverse signaling pathways. Plant J. 2003, 36, 602–615. [Google Scholar] [CrossRef]
- Navrot, N.; Collin, V.; Gualberto, J.; Gelhaye, E.; Hirasawa, M.; Rey, P.; Knaff, D.B.; Issakidis, E.; Jacquot, J.P.; Rouhier, N. Plant glutathione peroxidases are functional peroxiredoxins distributed in several subcellular compartments and regulated during biotic and abiotic stresses. Plant Physiol. 2006, 142, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Roxas, V.P.; Lodhi, S.A.; Garrett, D.K.; Mahan, J.R.; Allen, R.D. Stress tolerance in transgenic tobacco seedlings that overexpress glutathione S-transferase/glutathione peroxidase. Plant Cell Physiol. 2000, 41, 1229. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Vítámvás, P.; Prášil, I.T. The role of dehydrins in plant response to cold. Biol. Plant. 2007, 51, 601–617. [Google Scholar] [CrossRef]
- Tolleter, D.; Hincha, D.K.; Macherel, D.A. Mitochondrial late embryogenesis abundant protein stabilizes model membranes in the dry state. Biochim. Biophys. Acta 2010, 1798, 1926–1933. [Google Scholar] [CrossRef]
- Kawamura, Y.; Uemura, M. Mass spectrometric approach for identifying putative plasma membrane proteins of Arabidopsis leaves associated with cold acclimation. Plant J. 2003, 36, 141–154. [Google Scholar] [CrossRef]
- Lee, S.C.; Lee, M.Y.; Kim, S.J.; Jun, S.H.; An, G.; Kim, S.R. Characterization of an abiotic stress-inducible dehydrin gene, OsDhn1, in rice (Oryza sativa L.). Mol. Cells 2005, 19, 212–218. [Google Scholar] [PubMed]
- Peng, Y.H.; Reyes, J.L.; Wei, H.; Yang, Y.; Karlson, D.; Covarrubias, A.A.; Krebs, S.L.; Fessehaie, A.; Arora, R. RcDhn5, a cold acclimation–responsive dehydrin from Rhododendron catawbiense rescues enzyme activity from dehydration effects in vitro and enhances freezing tolerance in RcDhn5-overexpressing Arabidopsis plants. Physiol. Plant. 2008, 134, 583–597. [Google Scholar] [CrossRef]
- Yu, D.D.; Zhang, L.H.; Zhao, K.; Niu, R.X.; Zhai, H.; Zhang, J.X. VaERD15, a transcription factor gene associated with cold-tolerance in Chinese Wild Vitis amurensis. Front. Plant Sci. 2017, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, G.; Aldasoro, J.J. Activity of the pentose phosphate pathway and changes in nicotinamide nucleotide content during germination of seeds of Cicer arietinum L. J. Exp. Bot. 1979, 30, 1163–1170. [Google Scholar] [CrossRef]
- Dennis, D.T.; Blakeley, S.D. Carbohydrate metabolism. In Biochemistry & Molecular Biology of Plants; Buchanan, B.B., Gruissem, W., Eds.; American Society of Plant Physiologists: Rockville, MD, USA, 2000; pp. 652–654. [Google Scholar]
- Xiong, Y.Q.; Defraia, C.; Williams, D.; Zhang, X.D.; Mou, Z.L. Characterization of Arabidopsis 6-phosphogluconolactonase T-DNA insertion mutants reveals an essential role for the oxidative section of the plastidic pentose phosphate pathway in plant growth and development. Plant Cell Physiol. 2009, 50, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Miclet, E.; Stoven, V.; Michels, P.A.; Opperdoes, F.R.; Lallemand, J.Y.; Duffieux, F. NMR spectroscopic analysis of the first two steps of the pentose-phosphate pathway elucidates the role of 6-phosphogluconolactonase. J. Biol. Chem. 2001, 276, 34840–34846. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Z.; Lin, S.Z.; Guo, H.; Zhang, Z.Y.; Chen, X.Y. Functional analysis of PsG6PDH, a cytosolic glucose-6-phosphate dehydrogenase gene from Populus suaveolens, and its contribution to cold tolerance improvement in tobacco plants. Biotechnol. Lett. 2013, 35, 1509. [Google Scholar] [CrossRef] [PubMed]
- Bonaventure, G.; Salas, J.J.; Pollard, M.R.; Ohlrogge, J.B. Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell 2003, 15, 1020–1033. [Google Scholar] [CrossRef]
- Zheng, H.Q.; Rowland, O.; Kunst, L. Disruptions of the Arabidopsis enoyl-CoA reductase gene reveal an essential role for very-long-chain fatty acid synthesis in cell expansion during plant morphogenesis. Plant Cell 2005, 17, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Roudier, F.; Gissot, L.; Beaudoin, F.; Haslam, R.; Michaelson, L.; Marion, J.; Molino, D.; Lima, A.; Bach, L.; Morin, H.; et al. Very-long-chain fatty acids are involved in polar auxin transport and developmental patterning in Arabidopsis. Plant Cell 2010, 22, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Durrett, T.P.; Benning, C.; Ohlrogge, J. Plant triacylglycerols as feedstocks for the production of biofuels. Plant J. 2008, 54, 593–607. [Google Scholar] [CrossRef]
- Dyer, J.M.; Stymne, S.; Green, A.G.; Carlsson, A.S. High value oil from plants. Plant J. 2008, 54, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Gornicki, P.; Haselkorn, R. Wheat acetyl-CoA carboxylase. Plant Mol. Biol. 1993, 22, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Li, B.; Yang, J.H.; Sui, N.; Yang, X.M.; Meng, Q.W. Overexpression of tomato chloroplast omega-3 fatty acid desaturase gene alleviates the photoinhibition of photosystems 2 and 1 under chilling stress. Photosynthesis 2008, 46, 185–192. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D.; Carlson, M. The AMP-activated/SNF1 protein kinase subfamily: Metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 1998, 67, 821–855. [Google Scholar] [CrossRef]
- Rodrigues, A.; Adamo, M.; Crozet, P.; Margalha, L.; Confraria, A.; Martinho, C.; Elias, A.; Rabissi, A.; Lumbreras, V.; González-Guzmán, M.; et al. ABI1 and PP2CA phosphatases are negative regulators of Snf1-related protein kinase1 signaling in Arabidopsis. Plant Cell 2013, 25, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Purcell, P.C.; Smith, A.M.; Halford, N.G. Antisense expression of a sucrose non-fermenting-1-related protein kinase sequence in potato results in decreased expression of sucrose synthase in tubers and loss of sucrose-inducibility of sucrose synthase transcripts in leaves. Plant J. 1998, 14, 195–202. [Google Scholar] [CrossRef]
- Rosnoblet, C.; Aubry, C.; Leprince, O.; Vu, B.L.; Rogniaux, H.; Buitink, J. The regulatory gamma subunit SNF4b of the sucrose non-fermenting-related kinase complex is involved in longevity and stachyose accumulation during maturation of Medicago truncatula seeds. Plant J. 2007, 51, 47–59. [Google Scholar] [CrossRef]
- Bradford, K.J.; Downie, A.B.; Gee, O.H.; Alvarado, V.; Yang, H.; Dahal, P. Abscisic acid and gibberellin differentially regulate expression of genes of the SNF1-related kinase complex in tomato seeds. Plant Physiol. 2003, 132, 1560–1576. [Google Scholar] [CrossRef]
- Stone, J.M.; Walker, J.C. Plant protein kinase families and signal transduction. Plant Physiol. 1995, 108, 451–457. [Google Scholar] [CrossRef]
- Schenk, P.W.; Snaar-Jagalska, B.E. Signal perception and transduction: The role of protein kinases. Biochim. Biophys. Acta 1999, 1449, 1–24. [Google Scholar] [CrossRef]
- Yoshida, T.; Nishimura, N.; Kitahata, N.; Kuromori, T.; Ito, T.; Asami, T.; Shinozaki, K.; Hirayama, T. ABA-hypersensitive germination 3 encodes a protein phosphatase 2C (AtPP2CA) that strongly regulates abscisic acid signaling during germination among Arabidopsis protein phosphatase 2Cs. Plant Physiol. 2006, 140, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.L.; Liu, L.X.; Xiao, B.L.; Li, D.P.; Xing, X.; Kong, X.P.; Li, D.Q. Enhanced tolerance to low temperature in tobacco by over-expression of a new maize protein phosphatase 2C, ZmPP2C2. J. Plant Physiol. 2010, 167, 1307–1315. [Google Scholar] [CrossRef]
- Li, W.Y.F.; Shao, G.; Lam, H.M. Ectopic expression of GmPAP3 alleviates oxidative damage caused by salinity and osmotic stresses. New Phytol. 2010, 178, 80–91. [Google Scholar] [CrossRef]
- Baenagonzález, E.; Sheen, J. Convergent energy and stress signaling. Trends Plant Sci. 2008, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.K.; Sunkar, R. Gene regulation during cold stress acclimation in plants. Methods Mol. Biol. 2010, 639, 39–55. [Google Scholar] [PubMed]
- Moon, B.Y.; Higashi, S.; Gombos, Z.; Murata, N. Unsaturation of the membrane lipids of chloroplasts stabilizes the photosynthetic machinery against low-temperature photoinhibition in TG tobacco plants. Proc. Natl. Acad. Sci. USA 1995, 92, 6219–6233. [Google Scholar] [CrossRef] [PubMed]
- Baena-González, E.; Rolland, F.; Thevelein, J.M.; Sheen, J. A central integrator of transcription networks in plant stress and energy signalling. Nature 2007, 448, 938. [Google Scholar] [CrossRef]
- Li, J.Q.; Zhu, G.L.; Li, J.X.; Tian, F.D.; Zhang, C.H.; Wu, G.L.; Wang, J.W. Breeding of new castor variety Tongbi 5. Inn. Mong. Agric. Sci. Technol. 2004, 1, 10–11. [Google Scholar]
- Auld, D.L.; Bettis, B.L.; Crock, J.E.; Kephart, D. Planting date and temperature effects on germination and seed yield of Chickpea. Agron. J. 1988, 80, 909–914. [Google Scholar] [CrossRef]
- Wen, B.; Du, C.; Li, G.; Ghali, F.; Jones, A.R.; Käll, L.; Xu, S.; Zhou, R.; Ren, Z.; Feng, Q.; et al. IPeak: An open source tool to combine results from multiple MS/MS search engines. Proteomics 2015, 15, 2916–2920. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Zhou, R.; Feng, Q.; Wang, Q.; Wang, J.; Liu, S. IQuant: An automated pipeline for quantitative proteomics based on isobaric tags. Proteomics 2014, 14, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Pathway | Number of DAPS | p-value |
|---|---|---|---|
| 1 | Fatty acid biosynthesis | 5 | 0.016388 |
| 2 | Biotin metabolism | 3 | 0.017208 |
| 3 | Fatty acid metabolism | 6 | 0.031722 |
| 4 | Cyanoamino acid metabolism | 3 | 0.033034 |
| 5 | Ribosome | 12 | 0.044644 |
| Protein ID | Description | iTRAQ Ratio | p Value | qPCR Ratio | p Value | Trends a | |
|---|---|---|---|---|---|---|---|
| iTRAQ | qPCR | ||||||
| B9S696 | dehydrin Xero | 1.45 ± 0.133 | 0.004 | 4.52 ± 0.009 | 0.000 | + | + |
| B9RCA6 | GPX | 1.62 ± 0.136 | 0.001 | 2.30 ± 0.116 | 0.003 | + | + |
| B9RTR0 | LEA D-34 | 1.75 ± 0.138 | 0.000 | 5.34 ± 0.512 | 0.013 | + | + |
| B9SB19 | PP2C | 1.21 ± 0.114 | 0.029 | 6.40 ± 0.952 | 0.029 | + | + |
| Q41134 | KASII | 1.38 ± 0.207 | 0.023 | 2.76 ± 0.501 | 0.050 | + | + |
| B9SVJ9 | SnRK1 α catalytic subunit | 1.32 ± 0.162 | 0.022 | 3.85 ± 0.530 | 0.032 | + | + |
| B9S3Z7 | LEA D-34 | 1.30 ± 0.150 | 0.021 | 1.16 ± 0.117 | 0.232 | + | = |
| B9STQ5 | IF5A | 1.46 ± 0.279 | 0.032 | 0.42 ± 0.073 | 0.121 | + | = |
| Protein Accession | Fold Change | Accumulated | Description |
|---|---|---|---|
| B9SKD1 | 1.25 | Up | 60S ribosomal protein L3 |
| B9RMF8 | 1.21 | Up | Zn-dependent exopeptidases superfamily protein |
| B9S4D5 | 1.24 | Up | 40S ribosomal protein S26 |
| B9SKG4 | 1.27 | Up | 40S ribosomal protein S11 |
| B9SBM0 | 1.25 | Up | 60S ribosomal protein L9 |
| B9SIV4 | 1.35 | Up | 60S ribosomal protein L7a |
| B9R982 | 1.21 | Up | 40S ribosomal protein S9 |
| B9RG16 | 1.23 | Up | 40S ribosomal protein S27 |
| B9SYV4 | 1.23 | Up | 60S ribosomal protein L21 |
| B9RQ66 | 1.43 | Up | 60S ribosomal protein L28 |
| B9SCT8 | 1.22 | Up | 60S ribosomal protein L10a |
| B9T040 | 1.5 | Up | 60S ribosomal protein L35 |
| Protein Accession | Fold Change | Accumulated | Description |
|---|---|---|---|
| Q41134 | 1.38 | Up | β-ketoacyl-acyl carrier protein synthase II |
| Q41135 | 1.45 | Up | β-ketoacyl-acyl carrier protein synthase I |
| B9S1E2 | 1.26 | Up | Biotin caboxylase (BC) subunit of Het-ACCase |
| B9TAH3 | 1.47 | Up | β-carboxyltransferase (β-CT) subunit of Het-ACCase |
| B9RF47 | 1.38 | Up | Short chain dehydrogenase |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, M.; Liu, X.; Zhang, L.; Duan, Q.; Zhang, J. Quantitative Proteomic Analysis of Castor (Ricinus communis L.) Seeds During Early Imbibition Provided Novel Insights into Cold Stress Response. Int. J. Mol. Sci. 2019, 20, 355. https://doi.org/10.3390/ijms20020355
Wang X, Li M, Liu X, Zhang L, Duan Q, Zhang J. Quantitative Proteomic Analysis of Castor (Ricinus communis L.) Seeds During Early Imbibition Provided Novel Insights into Cold Stress Response. International Journal of Molecular Sciences. 2019; 20(2):355. https://doi.org/10.3390/ijms20020355
Chicago/Turabian StyleWang, Xiaoyu, Min Li, Xuming Liu, Lixue Zhang, Qiong Duan, and Jixing Zhang. 2019. "Quantitative Proteomic Analysis of Castor (Ricinus communis L.) Seeds During Early Imbibition Provided Novel Insights into Cold Stress Response" International Journal of Molecular Sciences 20, no. 2: 355. https://doi.org/10.3390/ijms20020355
APA StyleWang, X., Li, M., Liu, X., Zhang, L., Duan, Q., & Zhang, J. (2019). Quantitative Proteomic Analysis of Castor (Ricinus communis L.) Seeds During Early Imbibition Provided Novel Insights into Cold Stress Response. International Journal of Molecular Sciences, 20(2), 355. https://doi.org/10.3390/ijms20020355