Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins




Abstract
1. Introduction
2. DNA Methylation Studies
2.1. 5-Cytosine Methylation and DNA Methyltransferases
2.2. DNA Methylation in Alzheimer’s Disease
3. miRNAs Epigenetic Effects
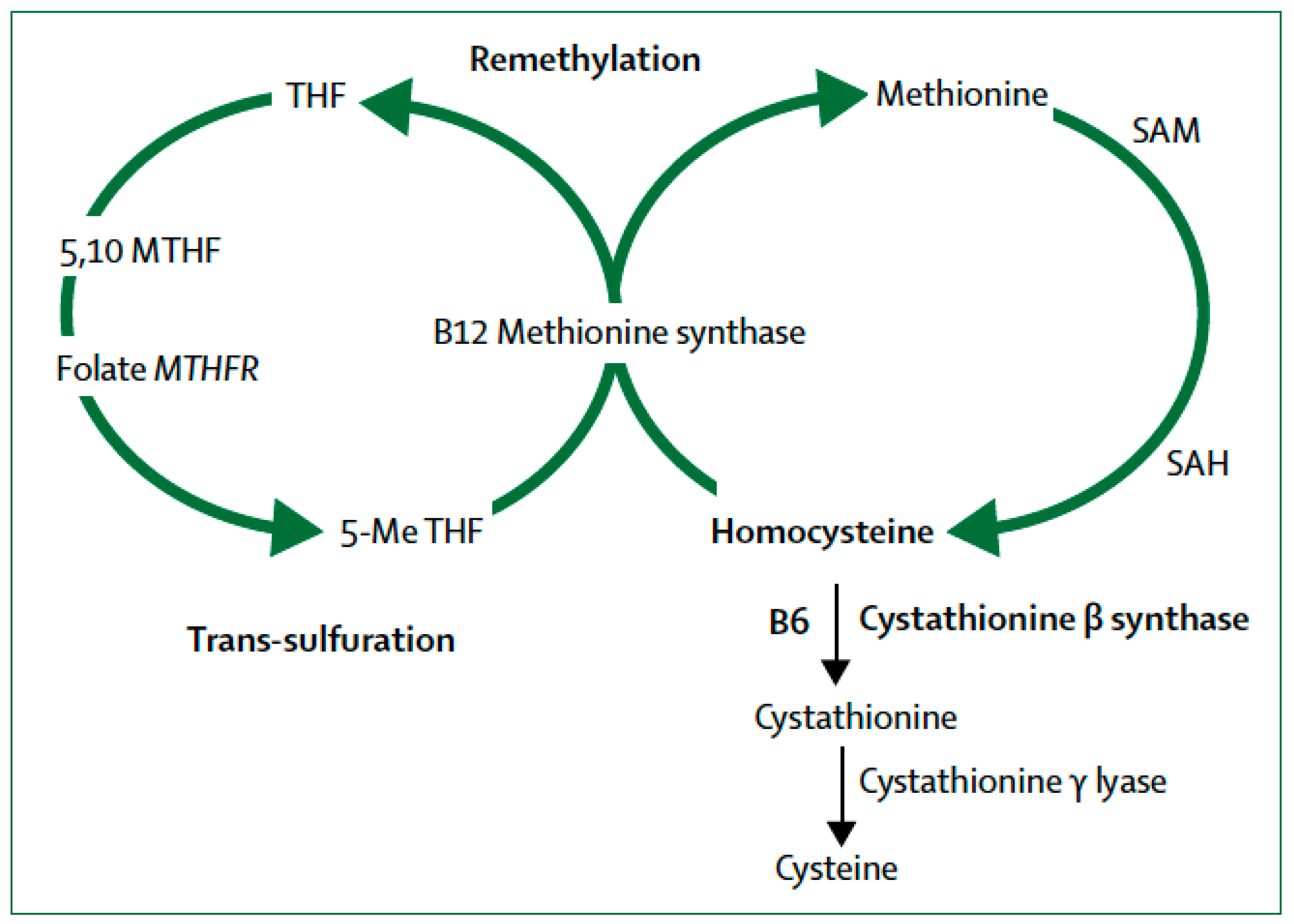
4. Transsulfuration Metabolic Pathways and Remethylation Defects
MTHFR and Epigenetic Drift
5. Homocysteine (Hcy): A Risk Factor for Cognitive Loss and Dementia
5.1. Hyperhomocysteinemia is an Independent Vascular Risk Factor
5.2. Genetic and Nongenetic Causes of Hyperhomocysteinemia
6. Folate Metabolism
Telomeres and Folate Levels
7. Vitamin B12 Deficiency and β-amyloid Deposition
7.1. Clinical Manifestations of Vitamin B12 Deficiency
7.2. Measuring Total Serum B12 Levels
7.3. Causes of Vitamin B12 Deficiency
7.4. Effects of B-Group Vitamins on Cognition: Negative Clinical Trials
8. SAM in Depression and Cognitive Loss
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roubroeks, J.A.Y.; Smith, R.G.; van den Hove, D.L.A.; Lunnon, K. Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J. Neurochem. 2017, 143, 158–170. [Google Scholar] [CrossRef]
- Millan, M.J. An epigenetic framework for neurodevelopmental disorders: From pathogenesis to potential therapy. Neuropharmacology 2013, 68, 2–82. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Pedersen, N.L.; Berg, S.; Johansson, B.; Johansson, K.; Mortimer, J.A.; Posner, S.F.; Viitanen, M.; Winblad, B.; Ahlbom, A. Heritability for Alzheimer’s disease: The study of dementia in Swedish twins. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, M117–M125. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef]
- Ma, X.; Liu, L.; Meng, J. MicroRNA-125b promotes neurons cell apoptosis and tau phosphorylation in Alzheimer’s disease. Neurosci. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Patrick, E.; Rajagopal, S.; Wong, H.A.; McCabe, C.; Xu, J.; Tang, A.; Imboywa, S.H.; Schneider, J.A.; Pochet, N.; Krichevsky, A.M.; et al. Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Irier, H.A.; Jin, P. Dynamics of DNA methylation in aging and Alzheimer’s disease. DNA Cell Biol. 2012, 31 (Suppl. 1), S42–S48. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C. MTHFR gene mutations: A potential marker of late-onset Alzheimer’s disease? J. Alzheimer’s Dis. 2015, 47, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huff, A.M.; Spence, J.D.; Hegele, R.A. Single nucleotide polymorphism in CTH associated with variation in plasma homocysteine concentration. Clin. Genet. 2004, 65, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.P.; Gavin, D.P.; Grayson, D.R. CpG methylation in neurons: Message, memory, or mask? Neuropsychopharmacology 2010, 35, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Hutnick, L.K.; Golshani, P.; Namihira, M.; Xue, Z.; Matynia, A.; Yang, X.W.; Silva, A.J.; Schweizer, F.E.; Fan, G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet. 2009, 18, 2875–2888. [Google Scholar] [CrossRef]
- Grossi, E.; Stoccoro, A.; Tannorella, P.; Migliore, L.; Coppedè, F. Artificial neural networks link one-carbon metabolism to gene-promoter methylation in Alzheimer’s disease. J. Alzheimers Dis. 2016, 53, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.Z.; Guan, W.P.; Maeda, T.; Makino, N. Analysis of telomere length and subtelomeric methylation of circulating leukocytes in women with Alzheimer’s disease. Aging Clin. Exp. Res. 2013, 25, 17–23. [Google Scholar] [CrossRef]
- Piaceri, I.; Raspanti, B.; Tedde, A.; Bagnoli, S.; Sorbi, S.; Nacmias, B. Epigenetic modifications in Alzheimer’s disease: Cause or effect? J. Alzheimers Dis. 2015, 43, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Barrachina, M.; Ferrer, I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol. 2009, 68, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimers Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.; Lovell, M.A. Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 2013, 134, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Nagata, K.; Hatsuta, H.; Takuma, H.; Bundo, M.; Iwamoto, K.; Tamaoka, A.; Murayama, S.; Saido, T.; Tsuji, S. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum. Mol. Genet. 2014, 23, 648–656. [Google Scholar] [CrossRef]
- Humphries, C.E.; Kohli, M.A.; Nathanson, L.; Whitehead, P.; Beecham, G.; Martin, E.; Mash, D.C.; Pericak-Vance, M.A.; Gilbert, J. Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 977–987. [Google Scholar] [CrossRef]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef]
- Ellison, E.M.; Abner, E.L.; Lovell, M.A. Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer’s disease. J. Neurochem. 2017, 140, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging. 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Hernández, H.G.; Sandoval-Hernández, A.G.; Garrido-Gil, P.; Labandeira-Garcia, J.L.; Zelaya, M.V.; Bayon, G.F.; Fernández, A.F.; Fraga, M.F.; Arboleda, G.; Arboleda, H. Alzheimer’s disease DNA methylome of pyramidal layers in frontal cortex: Laser-assisted microdissection study. Epigenomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; van den Hove, D.L.A.; Havermans, M.; van Casteren, A.; Le, K.X.; Palmour, R.; Lemere, C.A.; Rutten, B.P.F. Age-related epigenetic changes in hippocampal subregions of four animal models of Alzheimer’s disease. Mol. Cell. Neurosci. 2018, 86, 1–15. [Google Scholar] [CrossRef] [PubMed]
- OMIM®. Online Mendelian Inheritance in Man® Cystathionine Gamma-Lyase; CTH. Available online: http://www.omim.org/entry/607657 (accessed on 12 December 2018).
- OMIM®. Online Mendelian Inheritance in Man® 5,10-Methylenetetrahydrofolate Reductase; MTHFR. Available online: http://www.omim.org/entry/607093 (accessed on 12 December 2018).
- Linnebank, M.; Popp, J.; Smulders, Y.; Smith, D.; Semmler, A.; Farkas, M.; Kulic, L.; Cvetanovska, G.; Blom, H.; Stoffel-Wagner, B.; et al. S-adenosylmethionine is decreased in the cerebrospinal fluid of patients with Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, S.H.; Herrmann, W.; Obeid, R. Genetic defects in folate and cobalamin pathways affecting the brain. Clin. Chem. Lab. Med. 2013, 51, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.S.; Conus, N.; Kaput, J. B vitamin polymorphisms and behavior: Evidence of associations with neurodevelopment, depression, schizophrenia, bipolar disorder and cognitive decline. Neurosci. Biobehav. Rev. 2014, 47, 307–320. [Google Scholar] [CrossRef]
- Seripa, D.; Forno, G.D.; Matera, M.G.; Gravina, C.; Margaglione, M.; Palermo, M.T.; Wekstein, D.R.; Antuono, P.; Davis, D.G.; Daniele, A.; et al. Methylenetetrahydrofolate reductase, angiotensin converting enzyme gene polymorphisms in two genetically, and diagnostically distinct cohort of Alzheimer patients. Neurobiol. Aging 2003, 24, 933–939. [Google Scholar] [CrossRef]
- Da Silva, V.C.; da Costa Ramos, F.J.; Malaquias Freitas, E.; de Brito-Marques, P.R.; de Holanda Cavalcanti, M.N.; D’Almeida, V.; Cabral-Filho, J.E.; Cartaxo Muniz, M.T. Alzheimer’s disease in Brazilian elderly has a relation with homocysteine but not with MTHFR polymorphisms. Arq. Neuro-Psiquiatr. 2006, 64, 941–945. [Google Scholar] [CrossRef]
- Nishiyama, M.; Kato, Y.; Hashimoto, M.; Yukawa, S.; Omori, K. Apolipoprotein E, methylenetetrahydrofolate reductase (MTHFR) mutation and the risk of senile dementia—An epidemiological study using the polymerase chain reaction (PCR) method. Epidemiology 2000, 10, 163–172. [Google Scholar] [CrossRef]
- Ford, A.H.; Flicker, L.; Alfonso, H.; Hankey, G.J.; Norman, P.E.; van Bockxmeer, F.M.; Almeida, O.P. Plasma homocysteine and MTHFRC667T polymorphism as risk factors for incident dementia. J. Neurol. Neurosurg. Psychiatry 2012, 83, 70–75. [Google Scholar] [CrossRef] [PubMed]
- De Lau, L.M.L.; van Meurs, J.B.; Uitterlinden, A.G.; Smith, A.D.; Refsum, H.; Johnston, C.; Breteler, M.M. Genetic variation in homocysteine metabolism, cognition, and white matter lesions. Neurobiol. Aging 2010, 31, 2020–2022. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, L.; Fekih-Mrissa, N.; Klai, S.; Mansour, M.; Gritli, N.; Mrissa, R. Association of methylenetetrahydrofolate reductase polymorphisms with susceptibility to Alzheimer’s disease. Clin. Neurol. Neurosurg. 2013, 115, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Martin, G.M. Epigenetic drift in aging identical twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10413–10414. [Google Scholar] [CrossRef]
- Cook, R.H.; Schneck, S.A.; Clark, D.B. Twins with Alzheimer’s disease. Arch. Neurol. 1981, 38, 300–301. [Google Scholar] [CrossRef]
- Breitner, J.C.; Gatz, M.; Bergem, A.L.; Christian, J.C.; Mortimer, J.A.; McClearn, G.E.; Heston, L.L.; Welsh, K.A.; Anthony, J.C.; Folstein, M.F. Use of twin cohorts for research in Alzheimer’s disease. Neurology 1993, 43, 261–267. [Google Scholar] [CrossRef]
- Nee, L.E.; Lippa, C.F. Alzheimer’s disease in 22 twin pairs—13-year follow-up: Hormonal, infectious and traumatic factors. Dement. Geriatr. Cogn. Disord. 1999, 10, 148–151. [Google Scholar] [CrossRef]
- Coppedè, F. One-carbon metabolism and Alzheimer’s disease: Focus on epigenetics. Curr. Genom. 2010, 11, 246–260. [Google Scholar] [CrossRef]
- Spence, J.D.; Yi, Q.; Hankey, G.J. B vitamins in stroke prevention: Time to reconsider. Lancet Neurol. 2017, 16, 750–760. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H. Homocysteine, B vitamins, and cognitive impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.L.; Qiao, N.; Scott, T.; Rosenberg, I.; Spiro, A. High homocysteine and low B vitamins predict cognitive decline in aging men: The Veterans Affairs Normative Aging Study. Am. J. Clin. Nutr. 2005, 82, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Lipnicki, D.M.; Sachdev, P.S.; Crawford, J.; Reppermund, S.; Kochan, N.A.; Trollor, J.N.; Draper, B.; Slavin, M.J.; Kang, K.; Lux, O.; et al. Risk factors for late-life cognitive decline and variation with age and sex in the Sydney Memory and Ageing Study. PLoS ONE 2013, 8, e65841. [Google Scholar] [CrossRef] [PubMed]
- Dufouil, C.; Alperovitch, A.; Ducros, V.; Tzourio, C. Homocysteine, white matter hyperintensities, and cognition in healthy elderly people. Ann. Neurol. 2003, 53, 214–221. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Hudson, P.; Davies, G.; Hughes, A.; Williams, J.H.; Wilkinson, C. Homocysteine and cognitive decline in healthy elderly. Dement. Geriatr. Cogn. Disord. 2001, 12, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Nurk, E.; Refsum, H.; Tell, G.S.; Engedal, K.; Vollset, S.E.; Ueland, P.M.; Nygaard, H.A.; Smith, A.D. Plasma total homocysteine and memory in the elderly: The Hordaland Homocysteine study. Ann. Neurol. 2005, 58, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Solomon, A.; Kåreholt, I.; Rusanen, M.; Hänninen, T.; Leiviskä, J.; Winblad, B.; Laatikainen, T.; Soininen, H.; Kivipelto, M. Associations between serum homocysteine, holotranscobalamin, folate and cognition in the elderly: A longitudinal study. J. Intern. Med. 2012, 271, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Birks, J.; Nexo, E.; Ueland, P.M.; Schneede, J.; Scott, J.; Molloy, A.; Evans, J.G. Low vitamin B-12 status and risk of cognitive decline in older adults. Am. J. Clin. Nutr. 2007, 86, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Mangialasche, F.; Kalpouzos, G.; Solomon, A.; Kåreholt, I.; Smith, A.D.; Refsum, H.; Wang, R.; Mühlmann, M.; Ertl-Wagner, B.; et al. Association of vitamin B12, folate, and sulfur amino acids with brain magnetic resonance imaging measures in older adults: A longitudinal population-based study. JAMA Psychiatry 2016, 73, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Solomon, A.; Kåreholt, I.; Leiviskä, J.; Rusanen, M.; Ahtiluoto, S.; Winblad, B.; Laatikainen, T.; Soininen, H.; Kivipelto, M. Homocysteine and holotranscobalamin and the risk of Alzheimer disease: A longitudinal study. Neurology 2010, 75, 1408–1414. [Google Scholar] [CrossRef]
- Faux, N.G.; Ellis, K.A.; Porter, L.; Fowler, C.J.; Laws, S.M.; Martins, R.N.; Pertile, K.K.; Rembach, A.; Rowe, C.C.; Rumble, R.L.; et al. Homocysteine, vitamin B12, and folic acid levels in Alzheimer’s disease, mild cognitive impairment, and healthy elderly: Baseline characteristics in subjects of the Australian Imaging Biomarker Lifestyle study. J. Alzheimers Dis. 2011, 27, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D. The worldwide challenge of the dementias: A role for B vitamins and homocysteine? Food Nutr. Bull. 2008, 29, S143–S172. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimers Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, H.; Watanabe, A.; Akatsu, H.; Adachi, K.; Ohtsuka, C.; Terayama, Y.; Hosono, T.; Takahashi, S.; Wakita, H.; Jung, C.G.; et al. Homocysteine, another risk factor for Alzheimer disease, impairs apolipoprotein E3 function. J. Biol. Chem. 2010, 285, 38382–38388. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, P.; Singh, N. Homocysteine excess: Delineating the possible mechanism of neurotoxicity and depression. Fundam. Clin. Pharmacol. 2015, 29, 522–528. [Google Scholar] [CrossRef]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef]
- Snowdon, D.A.; Tully, C.L.; Smith, C.D.; Riley, K.P.; Markesbery, W.R. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: Findings from the Nun study. Am. J. Clin. Nutr. 2000, 71, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Odegaard, A.; Thyagarajan, B.; Hayes, J.; Cruz, K.S.; Derosiers, M.F.; Tyas, S.L.; Gross, M.D. Blood folate is associated with asymptomatic or partially symptomatic Alzheimer’s disease in the Nun study. J. Alzheimers Dis. 2012, 28, 637–645. [Google Scholar] [CrossRef]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Nazki, F.H.; Sameer, A.S.; Ganaie, B.A. Folate: Metabolism, genes, polymorphisms and the associated diseases. Gene 2014, 533, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Panossian, L.A.; Porter, V.R.; Valenzuela, H.F.; Zhu, X.; Reback, E.; Masterman, D.; Cummings, J.L.; Effros, R.B. Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiol. Aging 2003, 24, 77–84. [Google Scholar] [CrossRef]
- Paul, L.; Cattaneo, M.; D’Angelo, A.; Sampietro, F.; Fermo, I.; Razzari, C.; Fontana, G.; Eugene, N.; Jacques, P.F.; Selhub, J. Telomere length in peripheral blood mononuclear cells is associated with folate status in men. J. Nutr. 2009, 139, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Choi, S.-W.; Girelli, D.; Mason, J.B.; Dolnikowski, G.G.; Bagley, P.J.; Olivieri, O.; Jacques, P.F.; Rosenberg, I.H.; Corrocher, R.; et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. USA 2002, 99, 5606–5811. [Google Scholar] [CrossRef] [PubMed]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Troesch, B.; Weber, P.; Mohajeri, M. Potential links between impaired one-carbon metabolism due to polymorphisms, inadequate B-vitamin status, and the development of Alzheimer’s disease. Nutrients 2016, 8, 803. [Google Scholar] [CrossRef]
- Religa, D.; Styczynska, M.; Peplonska, B.; Gabryelewicz, T.; Pfeffer, A.; Chodakowska, M.; Luczywek, E.; Wasiak, B.; Stepien, K.; Golebiowski, M.; et al. Homocysteine, apolipoprotein E and methylenetetrahydrofolate reductase in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2003, 16, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.D.; Warren, M.J.; Refsum, H. Chapter Six—Vitamin B12. Adv. Food Nutr. Res. 2018, 83, 215–279. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P. Vitamin B12 deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.; Scott, J.M.; Ueland, P.-M.; Cunningham, C.; Casey, M.; Molloy, A.M. Diagnostic accuracy of holotranscobalamin, methylmalonic acid, serum cobalamin, and other indicators of tissue vitamin B12 status in the elderly. Clin. Chem. 2011, 57, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Huang, L.K.; Lee, Y.T.; Chan, L.; Hong, C.T. Suboptimal baseline serum vitamin B12 is associated with cognitive decline in people with Alzheimer’s disease undergoing cholinesterase inhibitor treatment. Front. Neurol. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Haron, Y.; Evans, L.; Smith, M.; Freedman, M.; Román, G. Metabolic markers of cobalamin deficiency and cognitive function in normal older adults. J. Am. Geriatr. Soc. 2004, 52, 66–71. [Google Scholar] [CrossRef]
- Garcia, A.; Zanibbi, K. Homocysteine and cognitive function in elderly people. CMAJ 2004, 171, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Ruiz, P.J. Neurologic complications of Sjögren syndrome. MedLink Neurol. 2010, 11, 1034. [Google Scholar]
- Andrès, E.; Loukili, N.H.; Noel, E.; Kaltenbach, G.; Abdelgheni, M.B.; Perrin, A.E.; Noblet-Dick, M.; Maloisel, F.; Schlienger, J.L.; Blicklé, J.F. Vitamin B12 (cobalamin) deficiency in elderly patients. CMAJ 2004, 171, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Spence, D. Mechanisms of thrombogenesis in atrial fibrillation. Lancet 2009, 373, 1006. [Google Scholar] [CrossRef]
- Wallin, A.; Román, G.C.; Esiri, M.; Kettunen, P.; Svensson, J.; Paraskevas, G.P.; Kapaki, E. Update on vascular cognitive impairment associated with subcortical small-vessel disease. J. Alzheimers Dis. 2018, 62, 1417–1441. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Troesch, B.; Weber, P. Inadequate supply of vitamins and DHA in the elderly: Implications for brain aging and Alzheimer-type dementia. Nutrition 2015, 31, 261–275. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-β deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- McCleery, J.; Abraham, R.P.; Denton, D.A.; Rutjes, A.W.; Chong, L.Y.; Al-Assaf, A.S.; Griffith, D.J.; Rafeeq, S.; Yaman, H.; Malik, M.A.; et al. Vitamin and mineral supplementation for preventing dementia or delaying cognitive decline in people with mild cognitive impairment. Cochrane Database Syst. Rev. 2018, 11, CD011905. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment. A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef] [PubMed]
- Jager, C.A.; Oulhaj, A.; Jacoby, R.; Refsum, H.; Smith, A.D. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2012, 27, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Douauda, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. PNAS Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D. As Work Group of the American Psychiatric Association Council on Research S-Adenosylmethionine (SAMe) for Neuropsychiatric Disorders: A Clinician-Oriented Review of Research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Guiraud, S.P.; Corthésy, J.; Da Silva, L.; Migliavacca, E.; Tautvydaitė, D.; Oikonomidi, A.; Moullet, B.; Henry, H.; Métairon, S.; et al. One-carbon metabolism, cognitive impairment and CSF measures of Alzheimer pathology: Homocysteine and beyond. Alzheimers. Res. Ther. 2017, 9, 43. [Google Scholar] [CrossRef]
- Galizia, I.; Oldani, L.; Macritchie, K.; Amari, E.; Dougall, D.; Jones, T.N.; Lam, R.W.; Massei, G.J.; Yatham, L.N.; Young, A.H. S-adenosyl methionine (SAMe) for depression in adults. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]


{kind=link}
| Proposed Mechanisms | |
|---|---|
| Vascular Mechanisms | |
| 1 | Impairs endothelial function reducing inducible NO synthase |
| 2 | NO-mediated endothelial dysfunction in brain vasculature |
| 3 | Causes a leaky blood-brain barrier |
| 4 | Induces thrombosis |
| 5 | Cerebrovascular ischemia leading to neuronal death and tau tangle deposition |
| 6 | Affects lipid metabolism increasing cholesterol synthesis |
| 7 | Reduces synthesis of apolipoprotein 1 |
| 8 | Causes cerebral amyloid angiopathy |
| Neuronal Mechanisms | |
| 1 | Direct activation of NMDA receptor causes excitotoxic neuronal death |
| 2 | Homocysteic acid and cysteine sulfinic acid activate NMDA receptor causing neuronal death by excitotoxicity |
| 3 | Oxidative stress induced by generating superoxide and reactive oxygen species |
| 4 | Decreased activity of antioxidant enzymes |
| 5 | Formation and deposition of β-amyloid |
| 6 | Potentiates neurotoxic effects of β-amyloid by itself or via homocysteic acid |
| 7 | Activates tau kinases, such as Cdk5, causing tau tangle deposition |
| 8 | Triggers the cell cycle in neurons, leading to tangle formation and cell death |
| 9 | Causes DNA damage, limits DNA repair, leading to apoptosis |
| 10 | Increases SAH inhibiting methylation reactions, such as DNA cytosine methylation in promoters for amyloid genes, causing epigenetic effects |
| 11 | Inhibits PP2A activity leading to tau tangle deposition |
| 12 | Inhibits methylation of phosphatidyletanolamine |
| 13 | Stimulates endoplasmic reticulum stress response leading to amyloid formation |
| 14 | Activates the immune system |
| 15 | Decreases SAM-dependent synthesis of catecholamines and other neurotransmitters |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Román, G.C.; Mancera-Páez, O.; Bernal, C. Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins. Int. J. Mol. Sci. 2019, 20, 319. https://doi.org/10.3390/ijms20020319
Román GC, Mancera-Páez O, Bernal C. Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins. International Journal of Molecular Sciences. 2019; 20(2):319. https://doi.org/10.3390/ijms20020319
Chicago/Turabian StyleRomán, Gustavo C., Oscar Mancera-Páez, and Camilo Bernal. 2019. "Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins" International Journal of Molecular Sciences 20, no. 2: 319. https://doi.org/10.3390/ijms20020319
APA StyleRomán, G. C., Mancera-Páez, O., & Bernal, C. (2019). Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins. International Journal of Molecular Sciences, 20(2), 319. https://doi.org/10.3390/ijms20020319