Hepatocyte Growth Factor: A Microenvironmental Resource for Leukemic Cell Growth




Abstract
1. Introduction
2. HGF and c-MET Expression and Function in Hematological Neoplasms
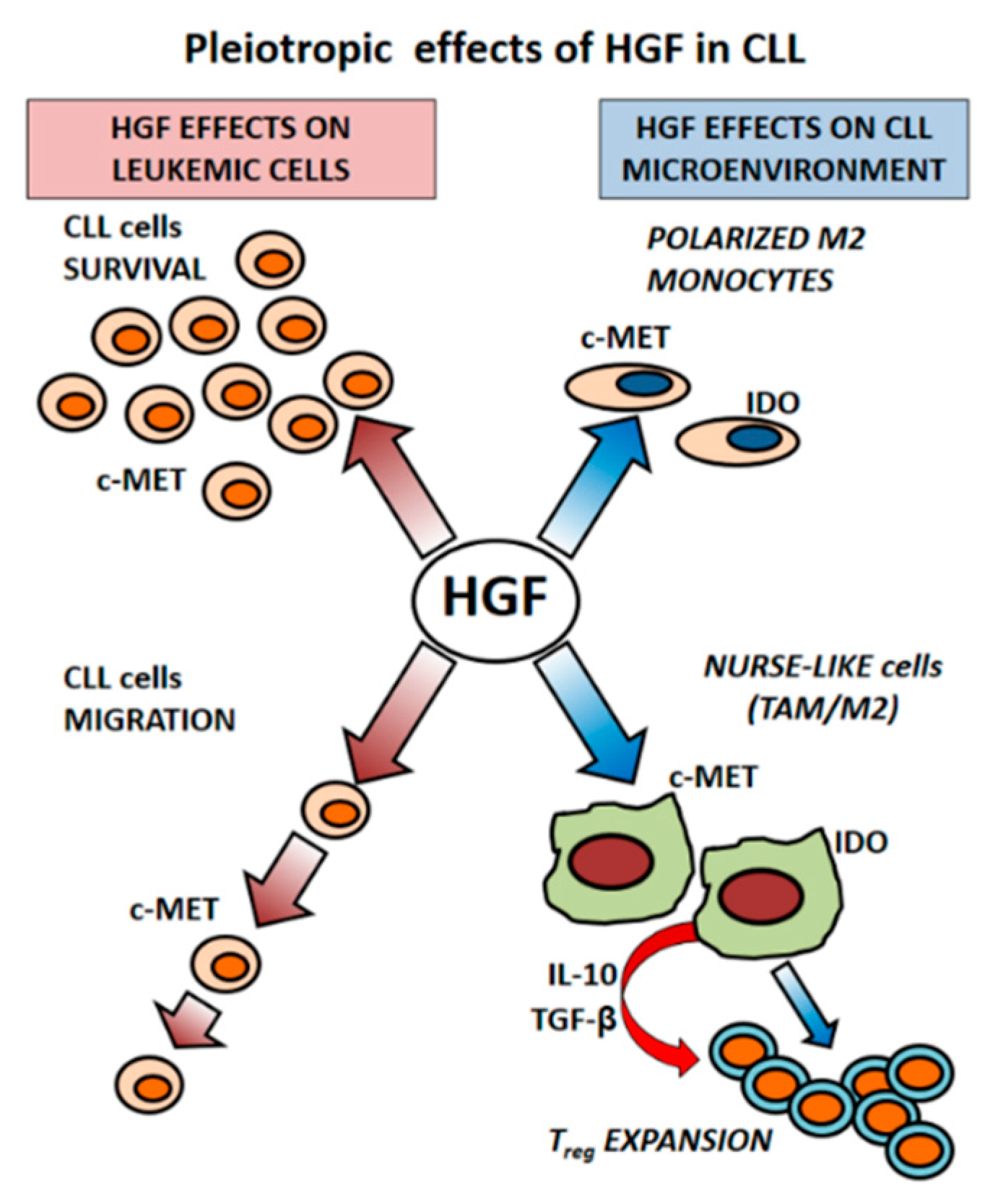
3. Microenvironment as a Source of HGF in CLL
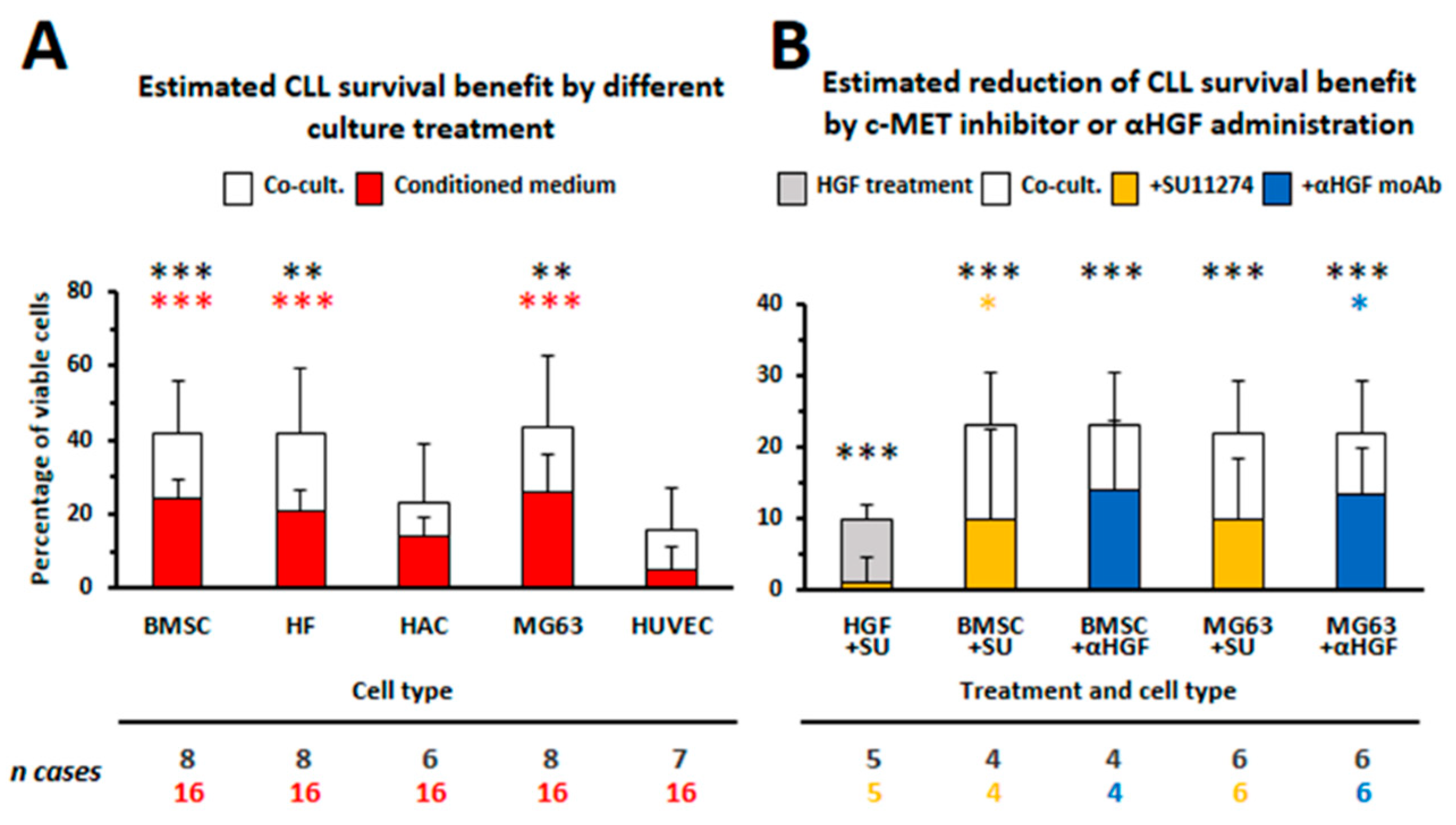
3.1. HGF Increases Viability of Leukemic Cells
3.2. CLL-Patient Monocytes Show Higher c-MET and IDO Expression than Controls Monocytes. HGF May Contribute to Drive Monocytes Toward an Immunosuppressive Phenotype
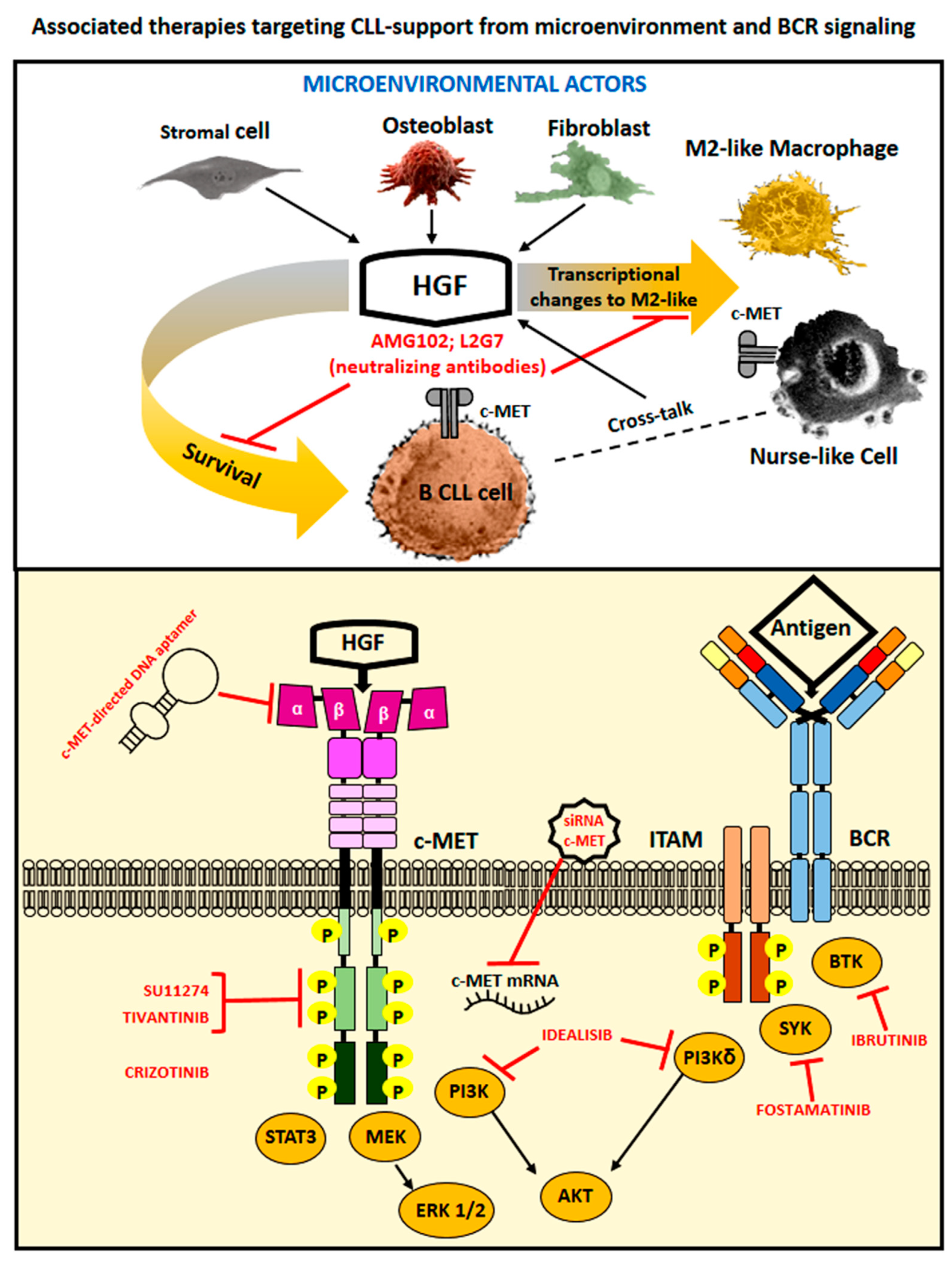
4. Therapeutic Options to Counteract HGF- and c-MET-Related Cancer Effects
5. Concluding Remarks
Author Contributions
Funding
Conflict of Interest
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; O’Brien, S. Evolution of CLL treatment—From chemoimmunotherapy to targeted and individualized therapy. Nat. Rev. Clin. Oncol. 2018, 15, 510–527. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Burger, J.A. Chronic lymphocytic leukemia therapy: New targeted therapies on the way. Expert Opin. Pharm. 2016, 17, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [PubMed]
- Caligaris-Cappio, F. Role of the microenvironment in chronic lymphocytic leukaemia. Br. J. Haematol. 2003, 123, 380–388. [Google Scholar] [CrossRef]
- Cutrona, G.; Tripodo, C.; Matis, S.; Recchia, A.G.; Massucco, C.; Fabbi, M.; Colombo, M.; Emionite, L.; Sangaletti, S.; Gulino, A.; et al. Microenvironmental regulation of the IL-23R/IL-23 axis overrides chronic lymphocytic leukemia indolence. Sci. Transl. Med. 2018, 10, eaal1571. [Google Scholar] [CrossRef] [PubMed]
- De Totero, D.; Meazza, R.; Capaia, M.; Fabbi, M.; Azzarone, B.; Balleari, E.; Gobbi, M.; Cutrona, G.; Ferrarini, M.; Ferrini, S. The opposite effects of IL-15 and IL-21 on CLL B cells correlate with differential activation of the JAK/STAT and ERK1/2 pathways. Blood 2008, 111, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, P.; de Totero, D. Is a protective microenvironment the Achille’s heel in chronic lymphocytic leukemia? In Frontiers in Clinical Drug Research-Hematology; Atta-ur-Rahman, Ed.; Bentham Science Publishers: Cambridge, UK, 2014; Volume 1, pp. 3–41. [Google Scholar]
- Lagneaux, L.; Delforge, A.; Bron, D.; De Bruyn, C.; Stryckmans, P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood 1998, 91, 2387–2396. [Google Scholar] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell. Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell. Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.Q.; Dai, L.; Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 2016, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Mahtouk, K.; Tjin, E.P.; Spaargaren, M.; Pals, S.T. The HGF/MET pathway as target for the treatment of multiple myeloma and B-cell lymphomas. Biochim. Biophys. Acta 2010, 1806, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Vilaine, M.; Hermouet, S. The Hepatocyte Growth Factor (HGF)/Met Axis: A Neglected Target in the Treatment of Chronic Myeloproliferative Neoplasms? Cancers 2014, 6, 1631–1669. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef]
- Gambella, M.; Palumbo, A.; Rocci, A. MET/HGF pathway in multiple myeloma: From diagnosis to targeted therapy? Expert Rev. Mol. Diagnos. 2015, 15, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Wader, K.F.; Fagerli, U.M.; Holt, R.U.; Borset, M.; Sundan, A.; Waage, A. Soluble c-Met in serum of patients with multiple myeloma: Correlation with clinical parameters. Eur. J. Haematol. 2011, 87, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Rocci, A.; Gambella, M.; Aschero, S.; Baldi, I.; Trusolino, L.; Cavallo, F.; Gay, F.; Larocca, A.; Magarotto, V.; Omede, P.; et al. MET dysregulation is a hallmark of aggressive disease in multiple myeloma patients. Br. J. Haematol. 2014, 164, 841–850. [Google Scholar] [CrossRef]
- Giannoni, P.; Scaglione, S.; Quarto, R.; Narcisi, R.; Parodi, M.; Balleari, E.; Barbieri, F.; Pattarozzi, A.; Florio, T.; Ferrini, S.; et al. An interaction between hepatocyte growth factor and its receptor (c-MET) prolongs the survival of chronic lymphocytic leukemic cells through STAT3 phosphorylation: A potential role of mesenchymal cells in the disease. Haematologica 2011, 96, 1015–1023. [Google Scholar] [CrossRef]
- Gohda, E.; Tsubouchi, H.; Nakayama, H.; Hirono, S.; Sakiyama, O.; Takahashi, K.; Miyazaki, H.; Hashimoto, S.; Daikuhara, Y. Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J. Clin. Investig. 1988, 81, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hiscox, S.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor/scatter factor, its molecular, cellular and clinical implications in cancer. Crit. Rev. Oncol./Hematol. 1999, 29, 209–248. [Google Scholar] [CrossRef]
- Van der Voort, R.; Taher, T.E.; Keehnen, R.M.; Smit, L.; Groenink, M.; Pals, S.T. Paracrine regulation of germinal center B cell adhesion through the c-met-hepatocyte growth factor/scatter factor pathway. J. Exp. Med. 1997, 185, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Reed, C.; Rice, K.L.; Sanda, T.; Rodig, S.J.; Tholouli, E.; Christie, A.; Valk, P.J.; Delwel, R.; Ngo, V.; et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Becker, M.W.; Tian, Q.; Lee, T.L.; Yan, X.; Liu, R.; Chiang, J.H.; Hood, L.; Clarke, M.F.; Weissman, I.L. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3396–3401. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Murota, H.; Kanda, S.; Hishikawa, Y.; Koji, T.; Taguchi, T.; Tanaka, Y.; Yamada, Y.; Ikeda, S.; Kohno, T.; et al. Expression of the c-Met proto-oncogene and its possible involvement in liver invasion in adult T-cell leukemia. Clin. Cancer Res. 2003, 9, 181–187. [Google Scholar] [PubMed]
- Onimaru, Y.; Tsukasaki, K.; Murata, K.; Imaizumi, Y.; Choi, Y.L.; Hasegawa, H.; Sugahara, K.; Yamada, Y.; Hayashi, T.; Nakashima, M.; et al. Autocrine and/or paracrine growth of aggressive ATLL cells caused by HGF and c-Met. Int. J. Oncol. 2008, 33, 697–703. [Google Scholar]
- Kumai, T.; Matsuda, Y.; Ohkuri, T.; Oikawa, K.; Ishibashi, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology 2015, 4, e976077. [Google Scholar] [CrossRef] [PubMed]
- Capello, D.; Gaidano, G.; Gallicchio, M.; Gloghini, A.; Medico, E.; Vivenza, D.; Buonaiuto, D.; Fassone, L.; Avanzi, G.C.; Saglio, G.; et al. The tyrosine kinase receptor met and its ligand HGF are co-expressed and functionally active in HHV-8 positive primary effusion lymphoma. Leukemia 2000, 14, 285–291. [Google Scholar] [CrossRef]
- Dai, L.; Trillo-Tinoco, J.; Cao, Y.; Bonstaff, K.; Doyle, L.; Del Valle, L.; Whitby, D.; Parsons, C.; Reiss, K.; Zabaleta, J.; et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. Blood 2015, 126, 2821–2831. [Google Scholar] [CrossRef]
- Lam, B.Q.; Dai, L.; Li, L.; Qiao, J.; Lin, Z.; Qin, Z. Molecular mechanisms of activating c-MET in KSHV+ primary effusion lymphoma. Oncotarget 2017, 8, 18373–18380. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Gunther, A.; Gradl, G.; Fonatsch, C.; Krueger, G.; Diehl, V.; Tesch, H. The Met/hepatocyte growth factor receptor (HGFR) gene is overexpressed in some cases of human leukemia and lymphoma. Leuk. Res. 1994, 18, 7–16. [Google Scholar] [CrossRef]
- Tjin, E.P.; Groen, R.W.; Vogelzang, I.; Derksen, P.W.; Klok, M.D.; Meijer, H.P.; van Eeden, S.; Pals, S.T.; Spaargaren, M. Functional analysis of HGF/MET signaling and aberrant HGF-activator expression in diffuse large B-cell lymphoma. Blood 2006, 107, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Kawano, R.; Ohshima, K.; Karube, K.; Yamaguchi, T.; Kohno, S.; Suzumiya, J.; Kikuchi, M.; Tamura, K. Prognostic significance of hepatocyte growth factor and c-MET expression in patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2004, 127, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.W.; Hwang, H.S.; Jung, S.J.; Park, C.; Yoon, D.H.; Suh, C.; Huh, J. Receptor tyrosine kinases MET and RON as prognostic factors in diffuse large B-cell lymphoma patients receiving R-CHOP. Cancer Sci. 2013, 104, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Al-Dayel, F.; Bu, R.; Bavi, P.; Al-Kuraya, K.S. Inhibition of c-MET is a potential therapeutic strategy for treatment of diffuse large B-cell lymphoma. Lab. Investig. 2010, 90, 1346–1356. [Google Scholar] [CrossRef]
- Bjorck, E.; Ek, S.; Landgren, O.; Jerkeman, M.; Ehinger, M.; Bjorkholm, M.; Borrebaeck, C.A.; Porwit-MacDonald, A.; Nordenskjold, M. High expression of cyclin B1 predicts a favorable outcome in patients with follicular lymphoma. Blood 2005, 105, 2908–2915. [Google Scholar] [CrossRef]
- Elenitoba-Johnson, K.S.; Jenson, S.D.; Abbott, R.T.; Palais, R.A.; Bohling, S.D.; Lin, Z.; Tripp, S.; Shami, P.J.; Wang, L.Y.; Coupland, R.W.; et al. Involvement of multiple signaling pathways in follicular lymphoma transformation: p38-mitogen-activated protein kinase as a target for therapy. Proc. Natl. Acad. Sci. USA 2003, 100, 7259–7264. [Google Scholar] [CrossRef]
- Giles, F.J.; Vose, J.M.; Do, K.A.; Johnson, M.M.; Manshouri, T.; Bociek, G.; Bierman, P.J.; O’Brien, S.M.; Kantarjian, H.M.; Armitage, J.O.; et al. Clinical relevance of circulating angiogenic factors in patients with non-Hodgkin’s lymphoma or Hodgkin’s lymphoma. Leuk. Res. 2004, 28, 595–604. [Google Scholar] [CrossRef]
- Hsiao, L.T.; Lin, J.T.; Yu, I.T.; Chiou, T.J.; Liu, J.H.; Yen, C.C.; Wang, W.S.; Chen, P.M. High serum hepatocyte growth factor level in patients with non-Hodgkin’s lymphoma. Eur. J. Haematol. 2003, 70, 282–289. [Google Scholar] [CrossRef]
- Seidel, C.; Borset, M.; Turesson, I.; Abildgaard, N.; Sundan, A.; Waage, A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood 1998, 91, 806–812. [Google Scholar] [PubMed]
- Wader, K.F.; Fagerli, U.M.; Holt, R.U.; Stordal, B.; Borset, M.; Sundan, A.; Waage, A. Elevated serum concentrations of activated hepatocyte growth factor activator in patients with multiple myeloma. Eur. J. Haematol. 2008, 81, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Pour, L.; Svachova, H.; Adam, Z.; Almasi, M.; Buresova, L.; Buchler, T.; Kovarova, L.; Nemec, P.; Penka, M.; Vorlicek, J.; et al. Levels of angiogenic factors in patients with multiple myeloma correlate with treatment response. Ann. Hematol. 2010, 89, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, I.B.; Christensen, J.H.; Lyng, M.B.; Moller, M.B.; Pedersen, L.; Rasmussen, L.M.; Ditzel, H.J.; Abildgaard, N. Hepatocyte growth factor pathway upregulation in the bone marrow microenvironment in multiple myeloma is associated with lytic bone disease. Br. J. Haematol. 2013, 161, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Standal, T.; Abildgaard, N.; Fagerli, U.M.; Stordal, B.; Hjertner, O.; Borset, M.; Sundan, A. HGF inhibits BMP-induced osteoblastogenesis: Possible implications for the bone disease of multiple myeloma. Blood 2007, 109, 3024–3030. [Google Scholar] [CrossRef]
- Borset, M.; Hjorth-Hansen, H.; Seidel, C.; Sundan, A.; Waage, A. Hepatocyte growth factor and its receptor c-met in multiple myeloma. Blood 1996, 88, 3998–4004. [Google Scholar]
- Borset, M.; Lien, E.; Espevik, T.; Helseth, E.; Waage, A.; Sundan, A. Concomitant expression of hepatocyte growth factor/scatter factor and the receptor c-MET in human myeloma cell lines. J. Biol. Chem. 1996, 271, 24655–24661. [Google Scholar] [CrossRef] [PubMed]
- Rampa, C.; Tian, E.; Vatsveen, T.K.; Buene, G.; Slordahl, T.S.; Borset, M.; Waage, A.; Sundan, A. Identification of the source of elevated hepatocyte growth factor levels in multiple myeloma patients. Biomark. Res. 2014, 2, 8. [Google Scholar] [CrossRef]
- Wader, K.F.; Fagerli, U.M.; Borset, M.; Lydersen, S.; Hov, H.; Sundan, A.; Bofin, A.; Waage, A. Immunohistochemical analysis of hepatocyte growth factor and c-Met in plasma cell disease. Histopathology 2012, 60, 443–451. [Google Scholar] [CrossRef]
- Pons, E.; Uphoff, C.C.; Drexler, H.G. Expression of hepatocyte growth factor and its receptor c-met in human leukemia-lymphoma cell lines. Leuk. Res. 1998, 22, 797–804. [Google Scholar] [CrossRef]
- Teofili, L.; Di Febo, A.L.; Pierconti, F.; Maggiano, N.; Bendandi, M.; Rutella, S.; Cingolani, A.; Di Renzo, N.; Musto, P.; Pileri, S.; et al. Expression of the c-met proto-oncogene and its ligand, hepatocyte growth factor, in Hodgkin disease. Blood 2001, 97, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Weimar, I.S.; de Jong, D.; Muller, E.J.; Nakamura, T.; van Gorp, J.M.; de Gast, G.C.; Gerritsen, W.R. Hepatocyte growth factor/scatter factor promotes adhesion of lymphoma cells to extracellular matrix molecules via alpha 4 beta 1 and alpha 5 beta 1 integrins. Blood 1997, 89, 990–1000. [Google Scholar] [PubMed]
- Bedewy, M.; El-Maghraby, S.; Bedewy, A. CD163 and c-Met expression in the lymph node and the correlations between elevated levels of serum free light chain and the different clinicopathological parameters of advanced classical Hodgkin’s lymphoma. Blood Res. 2013, 48, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Plattel, W.; van den Berg, A.; Ruther, N.; Huang, X.; Wang, M.; de Jong, D.; Vos, H.; van Imhoff, G.; Viardot, A.; et al. Expression of the c-Met oncogene by tumor cells predicts a favorable outcome in classical Hodgkin’s lymphoma. Haematologica 2012, 97, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, P.; Pietra, G.; Travaini, G.; Quarto, R.; Shyti, G.; Benelli, R.; Ottaggio, L.; Mingari, M.C.; Zupo, S.; Cutrona, G.; et al. Chronic Lymphocytic Leukemia Nurse-like cells express the hepatocyte growth factor receptor (c-MET) and indoleamine 2,3-dioxygenase and display features of immunosuppressive type 2 skewed macrophages. Haematologica 2014, 99, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef]
- Herndon, T.M.; Chen, S.S.; Saba, N.S.; Valdez, J.; Emson, C.; Gatmaitan, M.; Tian, X.; Hughes, T.E.; Sun, C.; Arthur, D.C.; et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia 2017, 31, 1340–1347. [Google Scholar] [CrossRef]
- Eksioglu-Demiralp, E.; Akdeniz, T.; Bayik, M. Aberrant expression of c-met and HGF/c-met pathway provides survival advantage in B-chronic lymphocytic leukemia. Cytom. Part B Clin. Cytom. 2010, 80, 1–7. [Google Scholar] [CrossRef]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar]
- Chen, P.M.; Liu, K.J.; Hsu, P.J.; Wei, C.F.; Bai, C.H.; Ho, L.J.; Sytwu, H.K.; Yen, B.L. Induction of immunomodulatory monocytes by human mesenchymal stem cell-derived hepatocyte growth factor through ERK1/2. J. Leukoc Biol. 2014, 96, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Bonanno, G.; Procoli, A.; Mariotti, A.; de Ritis, D.G.; Curti, A.; Danese, S.; Pessina, G.; Pandolfi, S.; Natoni, F.; et al. Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features. Blood 2006, 108, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [PubMed]
- Tsukada, N.; Burger, J.A.; Zvaifler, N.J.; Kipps, T.J. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 2002, 99, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, N.; Diener, S.; Idler, I.S.; Rauen, J.; Habe, S.; Busch, H.; Habermann, A.; Zenz, T.; Dohner, H.; Stilgenbauer, S.; et al. Nurse-like cells show deregulated expression of genes involved in immunocompetence. Br. J. Haematol. 2011, 154, 349–356. [Google Scholar] [CrossRef]
- Filip, A.A.; Cisel, B.; Koczkodaj, D.; Wasik-Szczepanek, E.; Piersiak, T.; Dmoszynska, A. Circulating microenvironment of CLL: Are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol. Dis. 2013, 50, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Bulgarelli, J.; Fiorcari, S.; Bertoncelli, L.; Martinelli, S.; Guarnotta, C.; Castelli, I.; Deaglio, S.; Debbia, G.; De Biasi, S.; et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013, 98, 1115–1123. [Google Scholar] [CrossRef]
- Ysebaert, L.; Fournie, J.J. Genomic and phenotypic characterization of nurse-like cells that promote drug resistance in chronic lymphocytic leukemia. Leuk. Lymphoma 2011, 52, 1404–1406. [Google Scholar] [CrossRef]
- Hanna, B.S.; McClanahan, F.; Yazdanparast, H.; Zaborsky, N.; Kalter, V.; Rossner, P.M.; Benner, A.; Durr, C.; Egle, A.; Gribben, J.G.; et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia 2016, 30, 570–579. [Google Scholar] [CrossRef]
- Galletti, G.; Caligaris-Cappio, F.; Bertilaccio, M.T. B cells and macrophages pursue a common path toward the development and progression of chronic lymphocytic leukemia. Leukemia 2016, 30, 2293–2301. [Google Scholar] [CrossRef]
- Galletti, G.; Scielzo, C.; Barbaglio, F.; Rodriguez, T.V.; Riba, M.; Lazarevic, D.; Cittaro, D.; Simonetti, G.; Ranghetti, P.; Scarfo, L.; et al. Targeting Macrophages Sensitizes Chronic Lymphocytic Leukemia to Apoptosis and Inhibits Disease Progression. Cell Rep. 2016, 14, 1748–1760. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Fedorchenko, O.; Rosen, N.; Koch, M.; Barthel, R.; Winarski, T.; Florin, A.; Wunderlich, F.T.; Reinart, N.; Hallek, M. LYN Kinase in the Tumor Microenvironment Is Essential for the Progression of Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, V.; Aittoniemi, J.; Jylhava, J.; Eklund, C.; Hurme, M.; Paavonen, T.; Oja, S.S.; Itala-Remes, M.; Sinisalo, M. Indoleamine 2,3-dioxygenase activity and expression in patients with chronic lymphocytic leukemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 363–365. [Google Scholar] [CrossRef] [PubMed]
- D'Arena, G.; Laurenti, L.; Minervini, M.M.; Deaglio, S.; Bonello, L.; De Martino, L.; De Padua, L.; Savino, L.; Tarnani, M.; De Feo, V.; et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk. Res. 2011, 35, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, K.; Schmitt, M.; Kowal, M.; Wlasiuk, P.; Bojarska-Junak, A.; Chen, J.; Rolinski, J.; Dmoszynska, A. Characterization of regulatory T cells in patients with B-cell chronic lymphocytic leukemia. Oncol. Rep. 2008, 20, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Yousefi, M.; Memarian, A.; Hojjat-Farsangi, M.; Khoshnoodi, J.; Razavi, S.M.; Jeddi-Tehrani, M.; Shokri, F. Increased frequency of CD8+ and CD4+ regulatory T cells in chronic lymphocytic leukemia: Association with disease progression. Cancer Investig. 2013, 31, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Jak, M.; Mous, R.; Remmerswaal, E.B.; Spijker, R.; Jaspers, A.; Yague, A.; Eldering, E.; Van Lier, R.A.; Van Oers, M.H. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Gordin, M.; Tesio, M.; Cohen, S.; Gore, Y.; Lantner, F.; Leng, L.; Bucala, R.; Shachar, I. c-Met and its ligand hepatocyte growth factor/scatter factor regulate mature B cell survival in a pathway induced by CD74. J. Immunol. 2010, 185, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Reinart, N.; Nguyen, P.H.; Boucas, J.; Rosen, N.; Kvasnicka, H.M.; Heukamp, L.; Rudolph, C.; Ristovska, V.; Velmans, T.; Mueller, C.; et al. Delayed development of chronic lymphocytic leukemia in the absence of macrophage migration inhibitory factor. Blood 2013, 121, 812–821. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Q.; Li, T.; Qian, J.; Lu, Y.; Li, Y.; Bi, E.; Reu, F.; Qin, Y.; Drazba, J.; et al. Role of Myeloma-Derived MIF in Myeloma Cell Adhesion to Bone Marrow and Chemotherapy Response. J. Natl. Cancer Inst. 2016, 108, djw131. [Google Scholar] [CrossRef]
- Piddock, R.E.; Marlein, C.R.; Abdul-Aziz, A.; Shafat, M.S.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. Myeloma-derived macrophage inhibitory factor regulates bone marrow stromal cell-derived IL-6 via c-MYC. J. Hematol. Oncol. 2018, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Pearce, L.; Morgan, L.; Robinson, S.; Ware, V.; Brennan, P.; Thomas, N.S.; Yallop, D.; Devereux, S.; Fegan, C.; et al. Mimicking the tumour microenvironment: Three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br. J. Haematol. 2012, 158, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Marrella, A.; Giannoni, P.; Pulsoni, I.; Quarto, R.; Raiteri, R.; Scaglione, S. Topographical Features of Graphene-Oxide-Functionalized Substrates Modulate Cancer and Healthy Cell Adhesion Based on the Cell Tissue of Origin. ACS Appl. Mater. Interfaces 2018, 10, 41978–41985. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Chiodoni, C.; Tripodo, C.; Colombo, M.P. The good and bad of targeting cancer-associated extracellular matrix. Curr. Opin. Pharmacol. 2017, 35, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Guerra, H.; Freitas, V.M. Extracellular Matrix Influencing HGF/c-MET Signaling Pathway: Impact on Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3300. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.R.; Kanninen, L.; Kaehr, B.; Townson, J.L.; Niklander, J.; Harjumaki, R.; Jeffrey Brinker, C.; Yliperttula, M. Silica bioreplication preserves three-dimensional spheroid structures of human pluripotent stem cells and HepG2 cells. Sci. Rep. 2015, 5, 13635. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef]
- Kato, T. Biological roles of hepatocyte growth factor-Met signaling from genetically modified animals. Biomed. Rep. 2017, 7, 495–503. [Google Scholar] [CrossRef]
- Baljevic, M.; Zaman, S.; Baladandayuthapani, V.; Lin, Y.H.; de Partovi, C.M.; Berkova, Z.; Amini, B.; Thomas, S.K.; Shah, J.J.; Weber, D.M.; et al. Phase II study of the c-MET inhibitor tivantinib (ARQ 197) in patients with relapsed or relapsed/refractory multiple myeloma. Ann. Hematol. 2017, 96, 977–985. [Google Scholar] [CrossRef]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, H.; Zhou, W.; Sun, S.; Zeng, Y.; Zhang, H.; Liang, L.; Xiao, X.; Song, J.; Ye, M.; et al. Targeting c-met receptor tyrosine kinase by the DNA aptamer SL1 as a potential novel therapeutic option for myeloma. J. Cell. Mol. Med. 2018, 22, 5978–5990. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, Y.; Yang, Y.; Li, A.; Bi, R.; Wang, L.; Shen, X.; Wang, W.; Jia, Y.; Yu, B.; et al. Activation of MET signaling by HDAC6 offers a rationale for a novel ricolinostat and crizotinib combinatorial therapeutic strategy in diffuse large B-cell lymphoma. J. Pathol. 2018, 246, 141–153. [Google Scholar] [CrossRef]
- Wu, K.; Xing, F.; Wu, S.Y.; Watabe, K. Extracellular vesicles as emerging targets in cancer: Recent development from bench to bedside. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 538–563. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Cao, H.; Shen, B.; Feng, J. Tumor-derived exosomes in cancer progression and treatment failure. Oncotarget 2015, 6, 37151–37168. [Google Scholar] [CrossRef]
- Lai, R.C.; Chen, T.S.; Lim, S.K. Mesenchymal stem cell exosome: A novel stem cell-based therapy for cardiovascular disease. Regener. Med. 2011, 6, 481–492. [Google Scholar] [CrossRef]
- Sdrimas, K.; Kourembanas, S. MSC microvesicles for the treatment of lung disease: A new paradigm for cell-free therapy. Antioxid. Redox signal. 2014, 21, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.G.; Feng, X.M.; Abbott, J.; Fang, X.H.; Hao, Q.; Monsel, A.; Qu, J.M.; Matthay, M.A.; Lee, J.W. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells 2014, 32, 116–125. [Google Scholar] [CrossRef]
- Riazifar, M.; Pone, E.J.; Lotvall, J.; Zhao, W. Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 125–154. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, X.; Tan, Y.; Li, Q.; Ma, J.; Wang, G. Mesenchymal Stem Cell Derived Exosomes in Cancer Progression, Metastasis and Drug Delivery: A Comprehensive Review. J. Cancer 2018, 9, 3129–3137. [Google Scholar] [CrossRef]
- Adachi, E.; Sakai, K.; Nishiuchi, T.; Imamura, R.; Sato, H.; Matsumoto, K. Different growth and metastatic phenotypes associated with a cell-intrinsic change of Met in metastatic melanoma. Oncotarget 2016, 7, 70779–70793. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Imanishi, S.; Azuma, K.; Kobayashi, C.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Replenishing exosomes from older bone marrow stromal cells with miR-340 inhibits myeloma-related angiogenesis. Blood Adv. 2017, 1, 812–823. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Neoplasia of Mature B Cells | Neoplastic Cells | c-MET Activation | Refs | |||
|---|---|---|---|---|---|---|
| c-MET | HGF | HGFA | Autocrine | Paracrine | ||
| Mantle Cell Lymphoma | − | − | n.d. | − | − | [34] |
| Marginal Zone Lymphoma | − | − | n.d. | − | − | [34] |
| Follicular Lymphoma | +/− | − | n.d. | − | + | [34] |
| Diffuse Large B Cell Lymphoma | + | − | + | − | + | [34,35] |
| Chronic Lymphocytic Leukemia | +/− | +/− | +/− | + | + | [21,34,56] |
| Burkitt Lymphoma | +/− | − | n.d. | − | + | [30,33,34] |
| Primary Effusion Lymphoma | + | + | + | + | + | [30] |
| Multiple Myeloma | + | + | + | + | + | [47,48,50] |
| Hodgkin Lymphoma | + | − | n.d. | − | + | [51,52,53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannoni, P.; Fais, F.; Cutrona, G.; Totero, D.d. Hepatocyte Growth Factor: A Microenvironmental Resource for Leukemic Cell Growth. Int. J. Mol. Sci. 2019, 20, 292. https://doi.org/10.3390/ijms20020292
Giannoni P, Fais F, Cutrona G, Totero Dd. Hepatocyte Growth Factor: A Microenvironmental Resource for Leukemic Cell Growth. International Journal of Molecular Sciences. 2019; 20(2):292. https://doi.org/10.3390/ijms20020292
Chicago/Turabian StyleGiannoni, Paolo, Franco Fais, Giovanna Cutrona, and Daniela de Totero. 2019. "Hepatocyte Growth Factor: A Microenvironmental Resource for Leukemic Cell Growth" International Journal of Molecular Sciences 20, no. 2: 292. https://doi.org/10.3390/ijms20020292
APA StyleGiannoni, P., Fais, F., Cutrona, G., & Totero, D. d. (2019). Hepatocyte Growth Factor: A Microenvironmental Resource for Leukemic Cell Growth. International Journal of Molecular Sciences, 20(2), 292. https://doi.org/10.3390/ijms20020292