IL-1β Inflammatory Cytokine-Induced TP63 Isoform ∆NP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells

 , and
, and 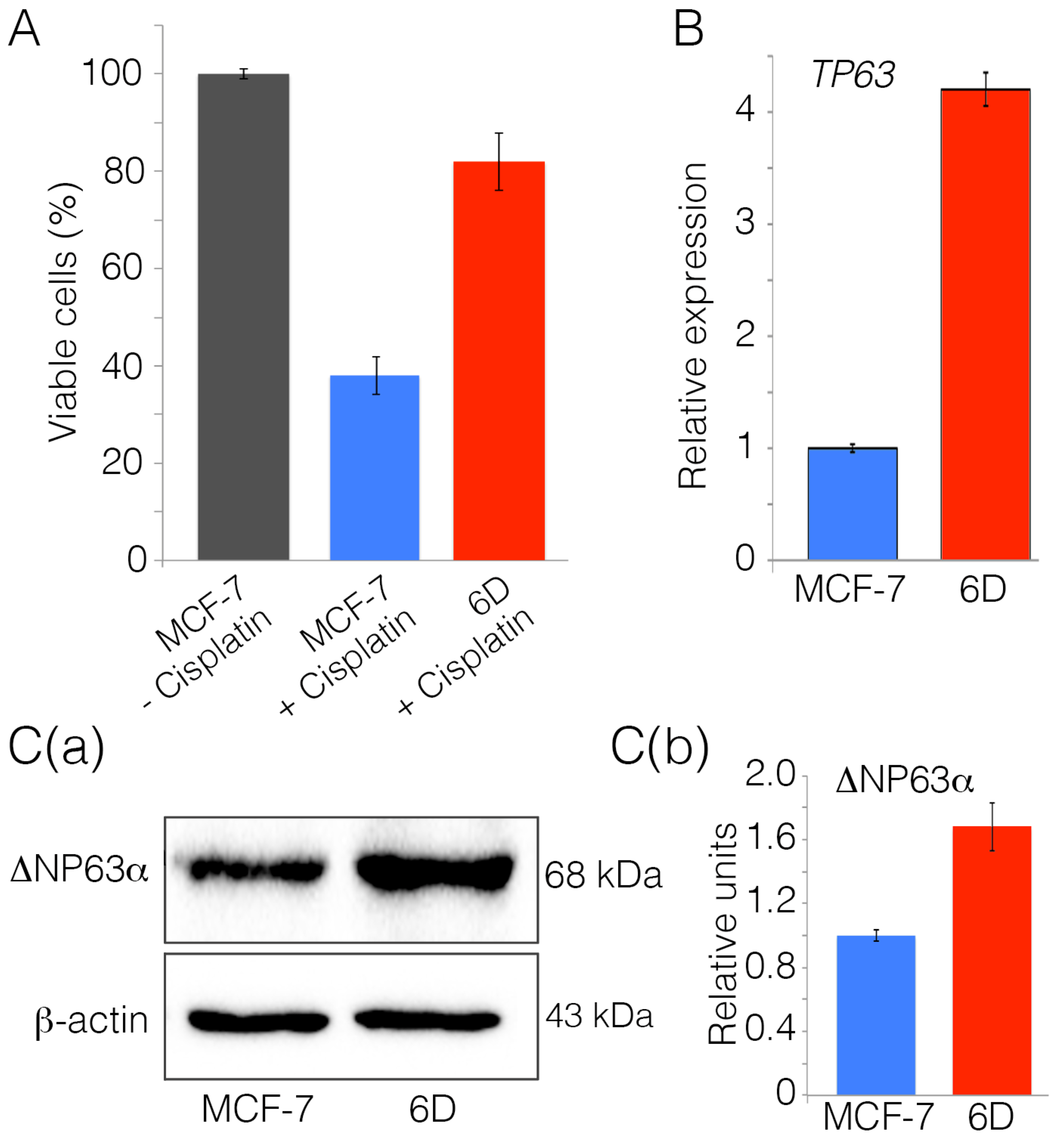{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Interleukin-1β Induces Upregulation of Chemoresistance-Related Genes in 6D Cells
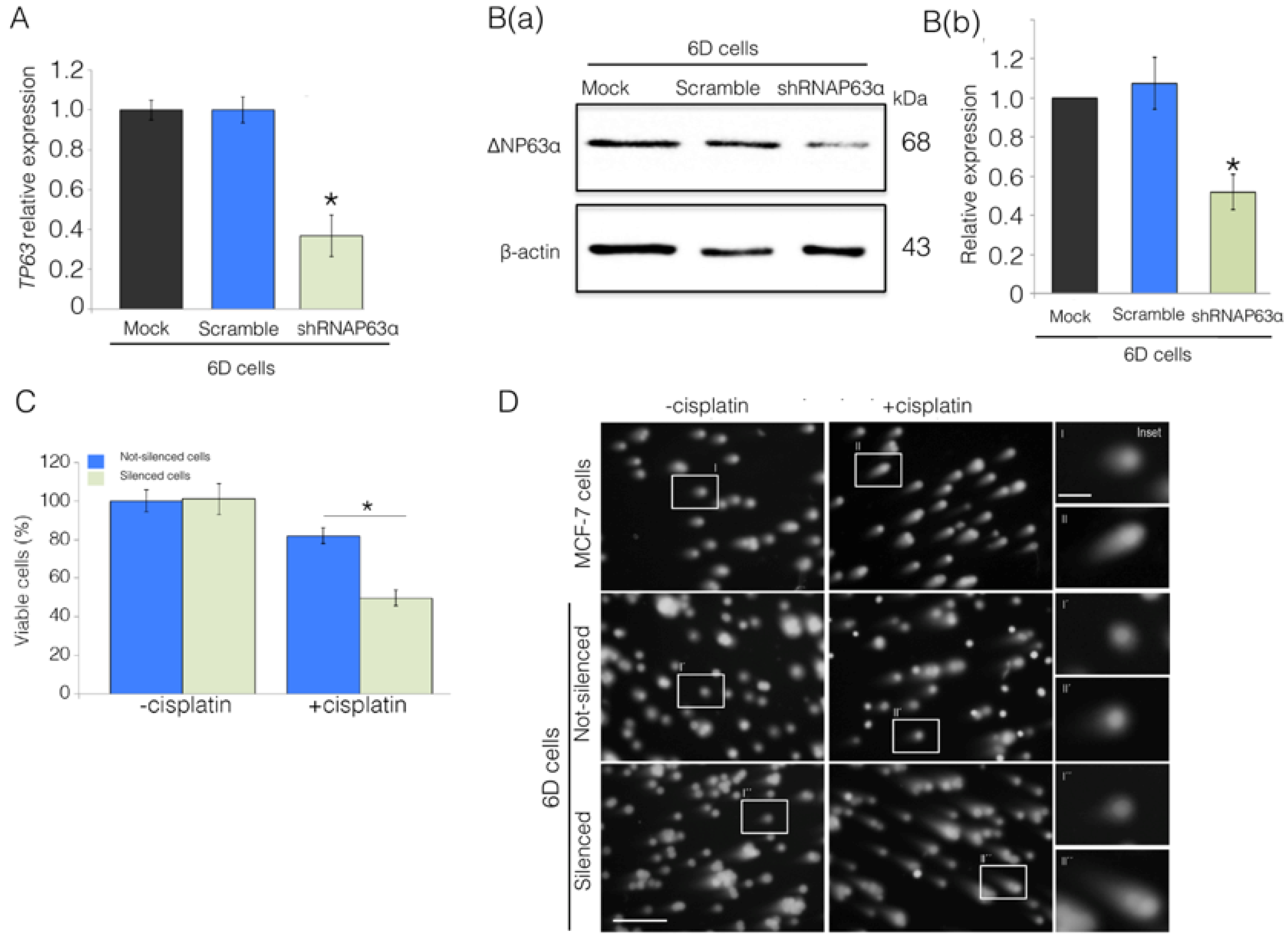
2.2. Silencing of ΔNp63α Decreases Resistance to Cisplatin in 6D Cells
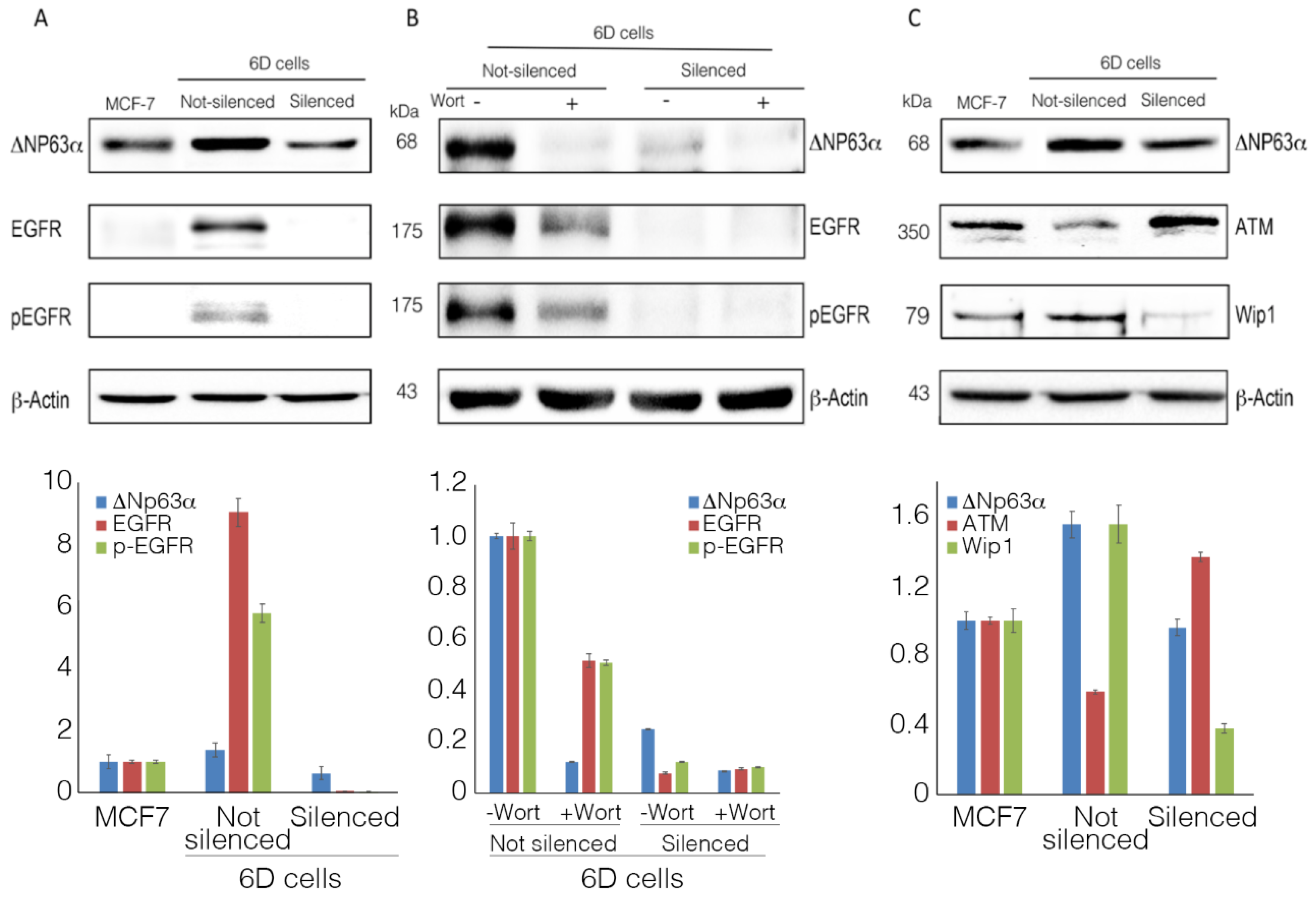
2.3. ΔNp63α Modulates the Expression and Activation of Survival Gene EGFR
2.4. ΔNp63α Modulates the Levels of Proteins ATM and Wip1
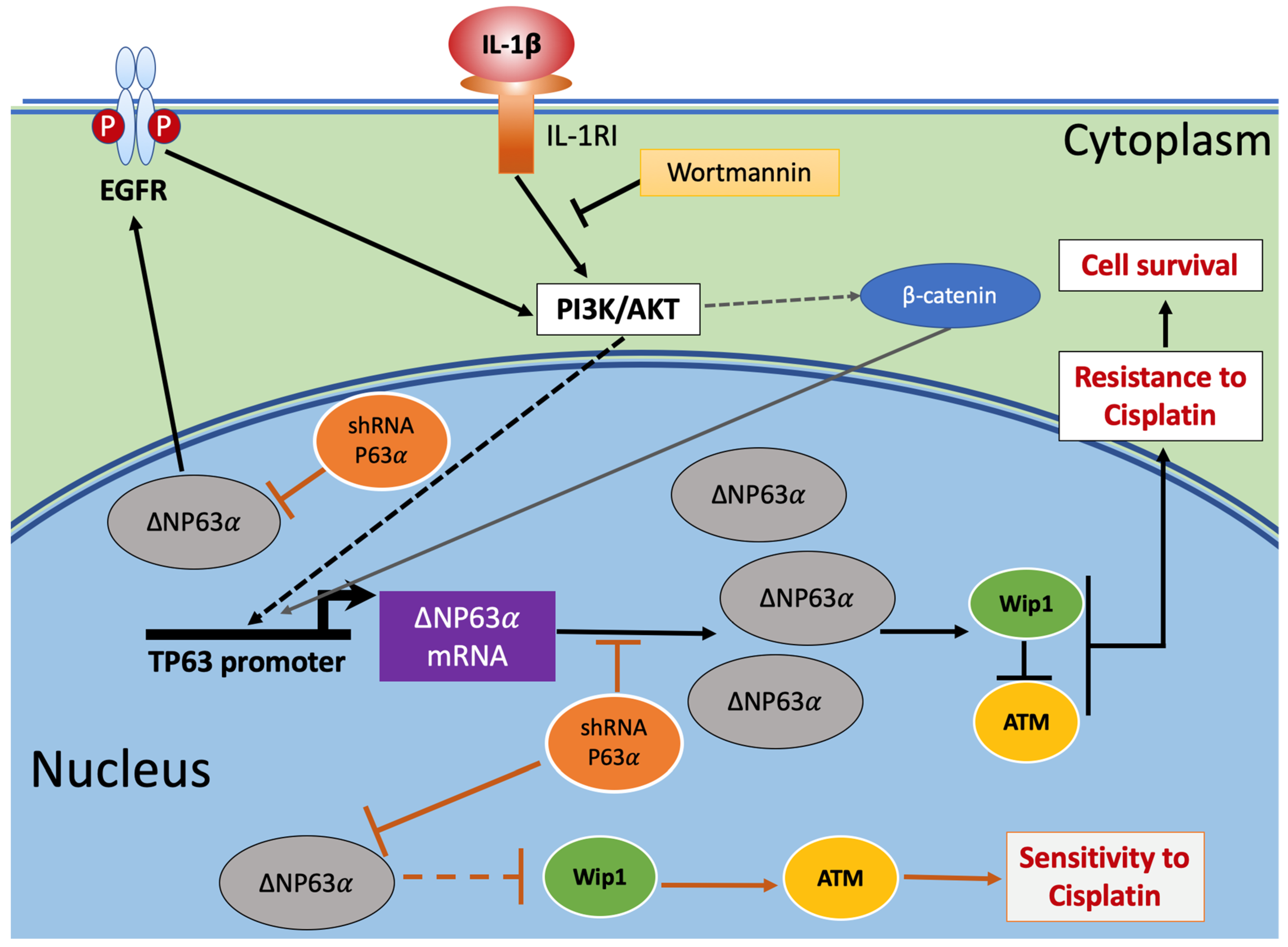
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Gene Expression
4.3. SDS-PAGE and Western Blotting
4.4. Resistance to Cisplatin
4.5. Silencing of 6D Cells With shRNAp63α
4.6. DNA Damage and Repair
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TP63 | Tumor protein p63 |
| ATM | Ataxia-telangiectasia mutated protein |
| Wip1 | Protein phosphatase 1D |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| MOI | Multiplicity of infection |
References
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Reveles-Espinoza, A.M.; Meza, I. Selection of a MCF-7 Breast Cancer Cell Subpopulation with High Sensitivity to IL-1beta: Characterization of and Correlation between Morphological and Molecular Changes Leading to Increased Invasiveness. Int. J. Breast Cancer 2012, 2012, 609148. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Lezama, R.; Meza, I. A novel beta-catenin signaling pathway activated by IL-1beta leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett. 2014, 354, 164–171. [Google Scholar] [CrossRef]
- Mendoza-Rodriguez, M.; Arevalo Romero, H.; Fuentes-Panana, E.M.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017, 390, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garduno, A.M.; Mendoza-Rodriguez, M.G.; Urrutia-Cabrera, D.; Dominguez-Robles, M.C.; Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induced methylation of the estrogen receptor ERalpha gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef]
- Meng, F.; Wu, G. Novel mechanism links p63 and cisplatin resistance. Cell Cycle 2014, 13, 881. [Google Scholar] [CrossRef] [PubMed]
- Vassilomanolakis, M.; Koumakis, G.; Barbounis, V.; Demiri, M.; Panopoulos, C.; Chrissohoou, M.; Apostolikas, N.; Efremidis, A.P. First-line chemotherapy with docetaxel and cisplatin in metastatic breast cancer. Breast 2005, 14, 136–141. [Google Scholar] [CrossRef]
- Perez, C.A.; Ott, J.; Mays, D.J.; Pietenpol, J.A. p63 consensus DNA-binding site: Identification, analysis and application into a p63MH algorithm. Oncogene 2007, 26, 7363–7370. [Google Scholar] [CrossRef]
- Huang, Y.; Sen, T.; Nagpal, J.; Upadhyay, S.; Trink, B.; Ratovitski, E.; Sidransky, D. ATM kinase is a master switch for the Delta Np63 alpha phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008, 7, 2846–2855. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Marchini, S.; Marabese, M.; Marrazzo, E.; Mariani, P.; Cattaneo, D.; Fossati, R.; Compagnoni, A.; Fruscio, R.; Lissoni, A.A.; Broggini, M. DeltaNp63 expression is associated with poor survival in ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2008, 19, 501–507. [Google Scholar] [CrossRef]
- Crook, T.; Nicholls, J.M.; Brooks, L.; O’Nions, J.; Allday, M.J. High level expression of deltaN-p63: A mechanism for the inactivation of p53 in undifferentiated nasopharyngeal carcinoma (NPC)? Oncogene 2000, 19, 3439–3444. [Google Scholar] [CrossRef]
- Ruptier, C.; De Gasperis, A.; Ansieau, S.; Granjon, A.; Taniere, P.; Lafosse, I.; Shi, H.; Petitjean, A.; Taranchon-Clermont, E.; Tribollet, V.; et al. TP63 P2 promoter functional analysis identifies beta-catenin as a key regulator of DeltaNp63 expression. Oncogene 2011, 30, 4656–4665. [Google Scholar] [CrossRef]
- Roman, S.; Petre, A.; Thepot, A.; Hautefeuille, A.; Scoazec, J.Y.; Mion, F.; Hainaut, P. Downregulation of p63 upon exposure to bile salts and acid in normal and cancer esophageal cells in culture. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G45–G53. [Google Scholar] [CrossRef]
- Sakaram, S.; Craig, M.P.; Hill, N.T.; Aljagthmi, A.; Garrido, C.; Paliy, O.; Bottomley, M.; Raymer, M.; Kadakia, M.P. Identification of novel DeltaNp63alpha-regulated miRNAs using an optimized small RNA-Seq analysis pipeline. Sci. Rep. 2018, 8, 10069. [Google Scholar] [CrossRef]
- Danilov, A.V.; Neupane, D.; Nagaraja, A.S.; Feofanova, E.V.; Humphries, L.A.; DiRenzo, J.; Korc, M. DeltaNp63alpha-mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PLoS ONE 2011, 6, e26815. [Google Scholar] [CrossRef]
- Grandis, J.R.; Sok, J.C. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol. Ther. 2004, 102, 37–46. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Hung, M.C. EGFR signaling pathway in breast cancers: From traditional signal transduction to direct nuclear translocalization. Breast Cancer Res. Treat. 2006, 95, 211–218. [Google Scholar] [CrossRef]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef]
- Delia, D.; Fontanella, E.; Ferrario, C.; Chessa, L.; Mizutani, S. DNA damage-induced cell-cycle phase regulation of p53 and p21waf1 in normal and ATM-defective cells. Oncogene 2003, 22, 7866–7869. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Sen, N.; Brait, M.; Begum, S.; Chatterjee, A.; Hoque, M.O.; Ratovitski, E.; Sidransky, D. DeltaNp63alpha confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res. 2011, 71, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Bretz, A.C.; Gittler, M.P.; Charles, J.P.; Gremke, N.; Eckhardt, I.; Mernberger, M.; Mandic, R.; Thomale, J.; Nist, A.; Wanzel, M.; et al. Delta Np63 activates the Fanconi anemia DNA repair pathway and limits the efficacy of cisplatin treatment in squamous cell carcinoma. Nucleic Acids Res. 2016, 44, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.Y.; Kim, J.Y.; Pelletier, J.F.; Vanderhyden, B.C.; Bachvarov, D.R.; Tsang, B.K. Akt confers cisplatin chemoresistance in human gynecological carcinoma cells by modulating PPM1D stability. Mol. Carcinog. 2015, 54, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Seed, B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003, 31, e154. [Google Scholar] [CrossRef] [PubMed]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Saharia, A.; Guittat, L.; Crocker, S.; Lim, A.; Steffen, M.; Kulkarni, S.; Stewart, S.A. Flap endonuclease 1 contributes to telomere stability. Curr. Biol. 2008, 18, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza-Rodríguez, M.G.; Ayala-Sumuano, J.T.; García-Morales, L.; Zamudio-Meza, H.; Pérez-Yepez, E.A.; Meza, I. IL-1β Inflammatory Cytokine-Induced TP63 Isoform ∆NP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 270. https://doi.org/10.3390/ijms20020270
Mendoza-Rodríguez MG, Ayala-Sumuano JT, García-Morales L, Zamudio-Meza H, Pérez-Yepez EA, Meza I. IL-1β Inflammatory Cytokine-Induced TP63 Isoform ∆NP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. International Journal of Molecular Sciences. 2019; 20(2):270. https://doi.org/10.3390/ijms20020270
Chicago/Turabian StyleMendoza-Rodríguez, Mónica G., Jorge T. Ayala-Sumuano, Lázaro García-Morales, Horacio Zamudio-Meza, Eloy A. Pérez-Yepez, and Isaura Meza. 2019. "IL-1β Inflammatory Cytokine-Induced TP63 Isoform ∆NP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells" International Journal of Molecular Sciences 20, no. 2: 270. https://doi.org/10.3390/ijms20020270
APA StyleMendoza-Rodríguez, M. G., Ayala-Sumuano, J. T., García-Morales, L., Zamudio-Meza, H., Pérez-Yepez, E. A., & Meza, I. (2019). IL-1β Inflammatory Cytokine-Induced TP63 Isoform ∆NP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. International Journal of Molecular Sciences, 20(2), 270. https://doi.org/10.3390/ijms20020270