Extracellular Matrix Alterations in Metastatic Processes



Abstract
1. Extracellular Matrix Structure and Function
2. The Metastatic Process
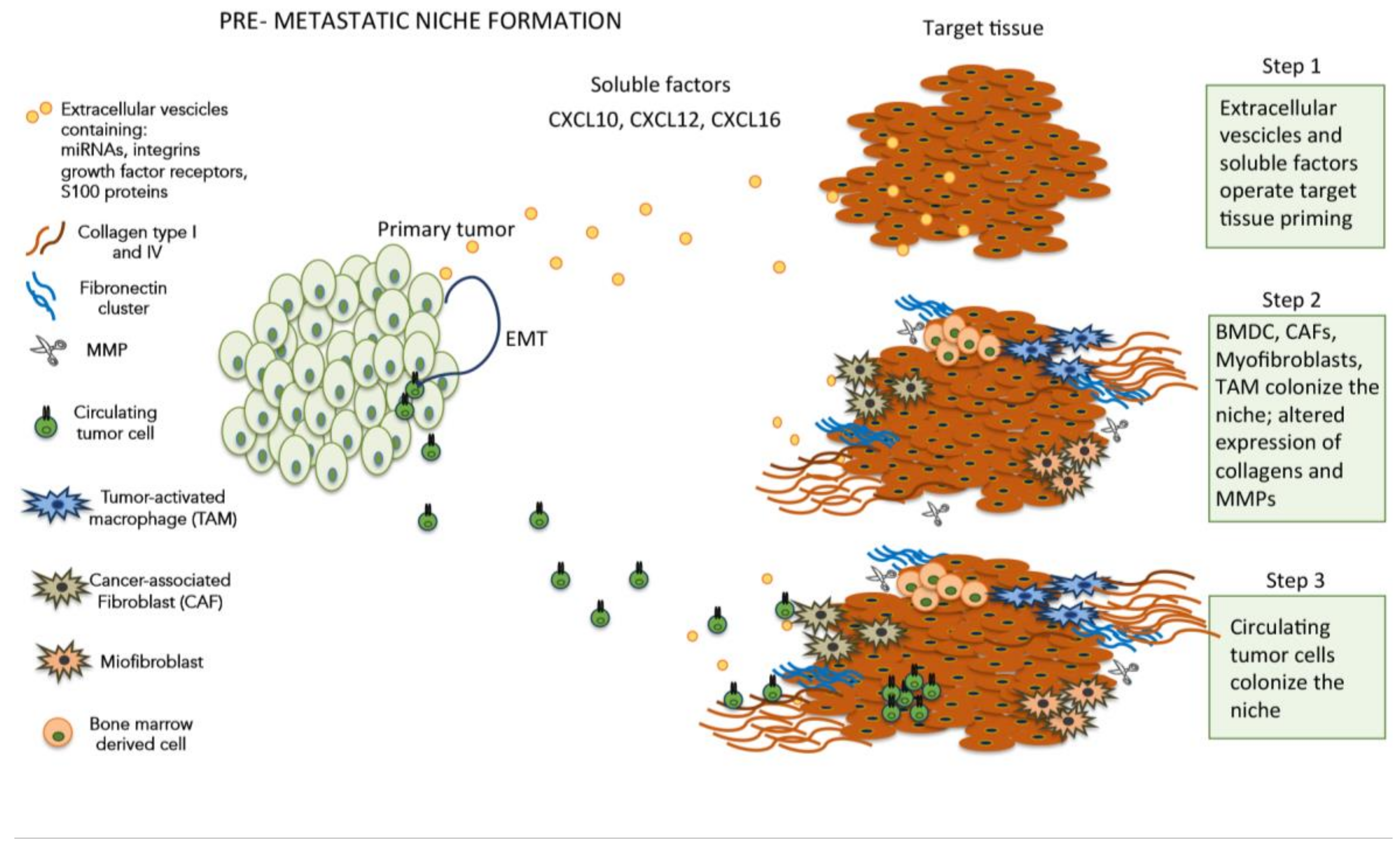
3. Premetastatic Niche and Tumor Microenvironment
4. The Tumor Microenvironment and ECM Modifications
4.1. Fibroblasts
4.2. Tumor Associate Macrophages (TAM)
4.3. Pericytes (PC)
4.4. Matrix Metallo Proteinases (MMPs)
5. Other Mechanisms Involved in ECM Alteration in Cancer Progression
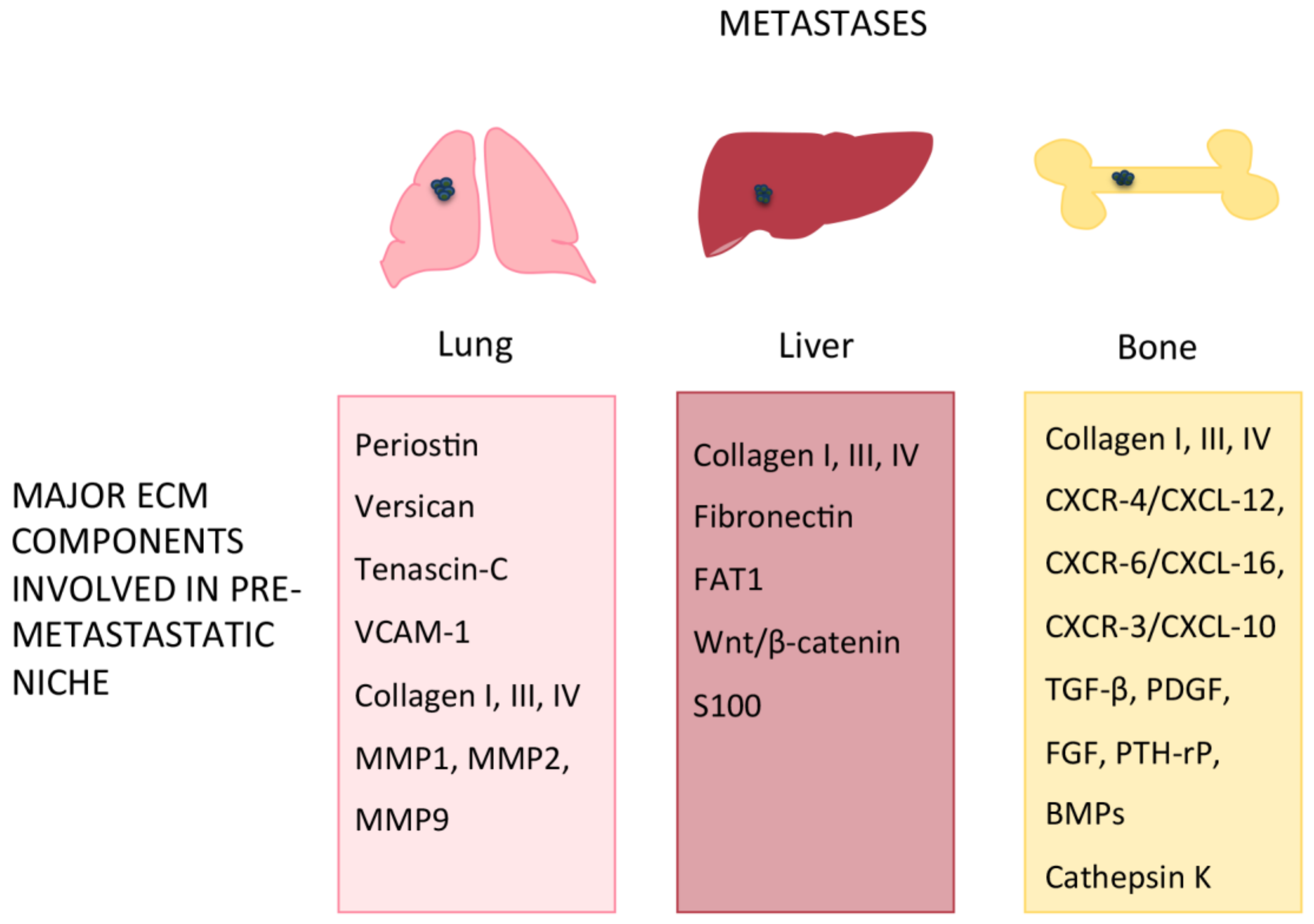
6. Organ-Specific Pre-Metastatic Niches
6.1. Lung
6.2. Liver
6.3. Bone
7. Conclusions
Funding
Conflicts of Interest
References
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Schaefer, L.; Schaefer, R.M. Proteoglycans: From structural compounds to signaling molecules. Cell Tissue Res. 2010, 339, 237–246. [Google Scholar] [CrossRef]
- Järveläinen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol. Rev. 2009, 61, 198–223. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.K.; Hahn, R.A. Collagens. Cell Tissue Res. 2010, 339, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Vindin, H.; Mithieux, S.M.; Weiss, A.S. Elastin architecture. Matrix Biol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Oxford, J.T.; Reeck, J.C.; Hardy, M.J. Extracellular Matrix in Development and Disease. Int. J. Mol. Sci. 2019, 20, 205. [Google Scholar] [CrossRef]
- Paolillo, M.; Schinelli, S. Integrins and exosomes, a dangerous liaison in cancer progression. Cancers 2017, 9, 95. [Google Scholar] [CrossRef]
- Paolillo, M.; Galiazzo, M.C.; Daga, A.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. An RGD small-molecule integrin antagonist induces detachment-mediated anoikis in glioma cancer stem cells. Int. J. Oncol. 2018, 53, 2683–2694. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, 99911. [Google Scholar] [CrossRef]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Lee, M.J.; Song, J.A.; Jeong, J.Y.; Kim, Y.K.; Lee, C.; Kim, T.H.; Kwak, K.B.; Kim, O.J.; An, H.J. Overexpression of goosecoid homeobox is associated with chemoresistance and poor prognosis in ovarian carcinoma. Oncol. Rep. 2014, 32, 189–198. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Chin, A.R.; Wang, S.E. Cancer tills the Premetastatic field: Mechanistic basis and clinical implications. Clin. Cancer Res. 2016, 22, 3725–3733. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-serum amyloid A3-TLR 4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef]
- Li, Y.; Kong, F.; Jin, C.; Hu, E.; Shao, Q.; Liu, J.; He, D.; Xiao, X. The expression of S100A8/S100A9 is inducible and regulated by the Hippo/YAP pathway in squamous cell carcinomas. BMC Cancer 2019, 19, 597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Høye, A.M.; Erler, J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016, 310, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a twist1-g3bp2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Sekiya, S.; Miura, S.; Matsuda-Ito, K.; Suzuki, A. Myofibroblasts Derived from Hepatic Progenitor Cells Create the Tumor Microenvironment. Stem Cell Rep. 2016, 7, 1130–1139. [Google Scholar] [CrossRef]
- Saini, F.; Argent, R.H.; Grabowska, A.M. Sonic Hedgehog Ligand: A Role in Formation of a Mesenchymal Niche in Human Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 424. [Google Scholar] [CrossRef]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016, 7, 6159–6174. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the tumor microenvironment: Shield or spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef] [PubMed]
- Chantrain, C.F.; Henriet, P.; Jodele, S.; Emonard, H.; Feron, O.; Courtoy, P.J.; DeClerck, Y.A.; Marbaix, E. Mechanisms of pericyte recruitment in tumour angiogenesis: A new role for metalloproteinases. Eur. J. Cancer 2006, 42, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.; Paulis, Y.W.; Nowak-Sliwinska, P.; Deumelandt, K.L.; Hosaka, K.; Soetekouw, P.M.; Cimpean, A.M.; Raica, M.; Pauwels, P.; van den Oord, J.J.; et al. Targeting PDGF-mediated recruitment of pericytes blocks vascular mimicry and tumor growth. J. Pathol. 2018, 246, 447–458. [Google Scholar] [CrossRef] [PubMed]
- You, W.K.; Stallcup, W.B. Localization of VEGF to Vascular ECM Is an Important Aspect of Tumor Angiogenesis. Cancers 2017, 9, 97. [Google Scholar] [CrossRef]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018, 115, 6410–6415. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Henriet, P.; Zhong, Z.D.; Brooks, P.C.; Weinberg, K.I.; DeClerck, Y.A. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27 KIP1. Proc. Natl. Acad. Sci. USA 2000, 97, 10026–10031. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Benzel, J.; Dorzweiler, K.; Cook, L.; Schlomann, U.; Zarbock, A.; Slater, E.P.; Nimsky, C.; Bartsch, J.W. ADAM8 in invasive cancers: Links to tumor progression, metastasis, and chemoresistance. Clin. Sci. (Lond.) 2019, 133, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, M.; Mineva, N.D.; Polmear, M.; Conrad, C.; Srinivasan, S.; Loussouarn, D.; Barillé-Nion, S.; Georgakoudi, I.; Dagg, Á.; McDermott, E.W.; et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol. Med. 2014, 6, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Götte, M.; Schlomann, U.; Roessler, M.; Pagenstecher, A.; Anderson, P.; Preston, J.; Pruessmeyer, J.; Ludwig, A.; Li, R.; et al. ADAM8 expression in breast cancer derived brain metastases: Functional implications on MMP-9 expression and transendothelial migration in breast cancer cells. Int. J. Cancer 2018, 142, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, D.; Sun, X.; Zhang, Y.; Wang, L.; Suo, J. ADAM17 promotes lymph node metastasis in gastric cancer via activation of the Notch and Wnt signaling pathways. Int. J. Mol. Med. 2019, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Geurts, N.; Becker-Pauly, C.; Martens, E.; Proost, P.; Van den Steen, P.E.; Stöcker, W.; Opdenakker, G. Meprins process matrix metalloproteinase-9 (MMP-9)/gelatinase B and enhance the activation kinetics by MMP-3. FEBS Lett. 2012, 586, 4264–4269. [Google Scholar] [CrossRef] [PubMed]
- Broder, C.; Becker-Pauly, C. The metalloproteases meprin α and meprin β: Unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem. J. 2013, 450, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Vadon-Le Goff, S.; Hulmes, D.J.; Moali, C. BMP-1/tolloid-like proteinases synchronize matrix assembly with growth factor activation to promote morphogenesis and tissue remodeling. Matrix Biol. 2015, 44–46, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, X.; Wang, T.; Xing, J. Overexpression of BMP1 reflects poor prognosis in clear cell renal cell carcinoma. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schröder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Mouw, J.K.; Yui, Y.; Damiano, L.; Bainer, R.O.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M.; et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360–367. [Google Scholar] [CrossRef]
- Tan, F.H.; Bai, Y.; Saintigny, P.; Darido, C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells 2019, 8, 333. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Díaz-Valdivia, N.I.; Calderón, C.C.; Díaz, J.E.; Lobos-González, L.; Sepulveda, H.; Ortíz, R.J.; Martinez, S.; Silva, V.; Maldonado, H.J.; Silva, P.; et al. Anti-neoplastic drugs increase caveolin-1-dependent migration, invasion and metastasis of cancer cells. Oncotarget 2017, 8, 111943–111965. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, J.; Klein, D. Caveolin-1, cancer and therapy resistance. Int. J. Cancer 2018, 143, 2092–2104. [Google Scholar] [CrossRef] [PubMed]
- Bourseau-Guilmain, E.; Menard, J.A.; Lindqvist, E.; Indira Chandran, V.; Christianson, H.C.; Cerezo Magaña, M.; Lidfeldt, J.; Marko-Varga, G.; Welinder, C.; Belting, M. Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat. Commun. 2016, 7, 11371. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.J.; Zhang, H.; Tang, G.S.; Wang, X.D.; Zheng, R.; Wang, Y.; Zhu, Y.; Xue, X.C.; Bi, J.W. Caveolin-1 is a modulator of fibroblast activation and a potential biomarker for gastric cancer. Int. J. Biol. Sci. 2015, 11, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Pixley, F.; Condeelis, J. Invadopodia and podosomes in tumor invasion. Eur. J. Cell Biol. 2006, 85, 213–218. [Google Scholar] [CrossRef]
- Clark, E.S.; Weaver, A.M. A new role for cortactin in invadopodia: Regulation of protease secretion. Eur. J. Cell Biol. 2008, 87, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Gong, H.; Yuan, Y.; Li, Y.; Wang, C.; Li, W.; Zhang, Z.; Liu, M.; Liu, H.; et al. miR-182 suppresses invadopodia formation and metastasis in non-small cell lung cancer by targeting cortactin gene. J. Exp. Clin. Cancer Res. 2018, 37, 141. [Google Scholar] [CrossRef]
- Soikkeli, J.; Podlasz, P.; Yin, M.; Nummela, P.; Jahkola, T.; Virolainen, S.; Krogerus, L.; Heikkilä, P.; von Smitten, K.; Saksela, O.; et al. Metastaticoutgrowth encompasses COL-I, FN1, and POSTN up-regulation and assembly to fibrillar networks regulating cell adhesion, migration, and growth. Am. J. Pathol. 2010, 177, 387–403. [Google Scholar] [CrossRef]
- Asano, K.; Nelson, C.M.; Nandadasa, S.; Aramaki-Hattori, N.; Lindner, D.J.; Alban, T.; Inagaki, J.; Ohtsuki, T.; Oohashi, T.; Apte, S.S.; et al. Stromal Versican Regulates Tumor Growth by Promoting Angiogenesis. Sci. Rep. 2017, 7, 17225. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4C stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martine, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Joshi, N.; Choi, H.; Ryu, S.; Hahn, M.; Catena, R.; Sadik, H.; Argani, P.; Wagner, P.; Vahdat, L.T.; et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012, 72, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Carlisle, R.; Ma, L.; Specian, R.D.; Gerritsen, M.E.; Granger, D.N. Endothelial expression of vascular cell adhesion molecule-1 correlates with metastatic pattern in spontaneous melanoma. Microcirculation 2001, 8, 335–345. [Google Scholar] [CrossRef]
- Amschler, K.; Kossmann, E.; Erpenbeck, L.; Kruss, S.; Schill, T.; Schön, M.; Möckel, S.M.C.; Spatz, J.P.; Schön, M.P. Nanoscale Tuning of VCAM-1 Determines VLA-4-Dependent Melanoma Cell Plasticity on RGD Motifs. Mol. Cancer Res. 2018, 16, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix metalloproteinases: Their functional role in lung cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef]
- Fanjul-Fernández, M.; Folgueras, A.R.; Fueyo, A.; Balbín, M.; Suárez, M.F.; Fernández-García, M.S.; Shapiro, S.D.; Freije, J.M.P.; López-Otín, C. Matrix metalloproteinase Mmp-1a is dispensable for normal growth and fertility in mice and promotes lung cancer progression by modulating inflammatory responses. J. Biol. Chem. 2018, 293, 11970. [Google Scholar] [CrossRef]
- Tsung, A.J.; Kargiotis, O.; Chetty, C.; Lakka, S.S.; Gujrati, M.; Spomar, D.G.; Dinh, D.H.; Rao, J.S. Downregulation of matrix metalloproteinase-2 (MMP-2) utilizing adenovirus-mediated transfer of small interfering RNA (siRNA) in a novel spinal metastatic melanoma model. Int. J. Oncol. 2008, 32, 557–564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.M.; Senior, R.M.; Shibuya, M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Zhong, Y.; Lu, Y.T.; Sun, Y.; Shi, Z.H.; Li, N.G.; Tang, Y.P.; Duan, J.A. Recent opportunities in matrix metalloproteinase inhibitor drug design for cancer. Expert Opin. Drug Discov. 2018, 13, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Williamson, T.; Sultanpuram, N.; Sendi, H. The role of liver microenvironment in hepatic metastasis. Clin. Transl. Med. 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Azizidoost, S.; Ahmadzadeh, A.; Rahim, F.; Shahjahani, M.; Seghatoleslami, M.; Saki, N. Hepatic metastatic niche: From normal to pre-metastatic and metastatic niche. Tumour Biol. 2016, 37, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Pecqueux, M.; Halama, N.; Dienemann, H.; Muley, T.; Pfannschmidt, J.; Lasitschka, F.; Klupp, F.; Schmidt, T.; Rahbari, N.; et al. Tumour-site-dependent expression profile of angiogenic factors in tumourassociated stroma of primary colorectal cancer and metastases. Br. J. Cancer 2014, 110, 441–449. [Google Scholar] [CrossRef]
- van Huizen, N.A.; Coebergh van den Braak, R.R.J.; Doukas, M.; Dekker, L.J.M.; IJzermans, J.N.M.; Luider, T.M. Up-regulation of collagen proteins in colorectal liver metastasis compared with normal liver tissue. J. Biol. Chem. 2019, 294, 281–289. [Google Scholar] [CrossRef]
- Barbazán, J.; Alonso-Alconada, L.; Elkhatib, N.; Geraldo, S.; Gurchenkov, V.; Glentis, A.; van Niel, G.; Palmulli, R.; Fernández, B.; Viaño, P.; et al. Liver Metastasis Is Facilitated by the Adherence of Circulating Tumor Cells to Vascular Fibronectin Deposits. Cancer Res. 2017, 77, 3431–3441. [Google Scholar] [CrossRef]
- Zenitani, M.; Nojiri, T.; Hosoda, H.; Kimura, T.; Uehara, S.; Miyazato, M.; Okuyama, H.; Kangawa, K. Chemotherapy can promote liver metastasis by enhancing metastatic niche formation in mice. J. Surg. Res. 2018, 224, 50–57. [Google Scholar] [CrossRef]
- Burnier, J.V.; Wang, N.; Michel, R.P.; Hassanain, M.; Li, S.; Lu, Y.; Metrakos, P.; Antecka, E.; Burnier, M.N.; Ponton, A.; et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene 2011, 30, 3766–3783. [Google Scholar] [CrossRef]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef] [PubMed]
- Lustig, B.; Behrens, J. The Wnt signaling pathway and its role in tumor development. J. Cancer Res. Clin. Oncol. 2003, 129, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, Ş.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, H.H.; Morgan, E.F.; Niebur, G.L.; Morris, G.E.; Wong, E.K.; Keaveny, T.M. Comparison of the elastic and yield properties of human femoral trabecular and cortical bone tissue. J. Biomech. 2004, 37, 27–35. [Google Scholar] [CrossRef]
- Kolb, A.D.; Bussard, K.M. The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers 2019, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2018, 15, 100205. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Brown, J.; Coleman, R. The role of biomarkers in the management of bone-homing malignancies. J. Bone Oncol. 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Rao, S.; Cronin, S.J.F.; Sigl, V.; Penninger, J.M. RANKL and RANK: From Mammalian Physiology to Cancer Treatment. Trends Cell Biol. 2018, 28, 213–223. [Google Scholar] [CrossRef]
- Matsugaki, A.; Harada, T.; Kimura, Y.; Sekita, A.; Nakano, T. Dynamic collision behavior between osteoblasts and tumor cells regulates the disordered arrangement of collagen fiber/apatite crystals in metastasized bone. Int. J. Mol. Sci. 2018, 19, 3474. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Hsia, S.M.; Shieh, T.M. Lysyl Oxidase and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Vargas, G.; Pape, F.L.; Clézardin, P. Cancer Cell colonisation in the bone microenvironment. Int. J. Mol. Sci. 2016, 17, 1674. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J. Role of Tumor-Derived Chemokines in Osteolytic Bone Metastasis. Front. Endocrinol. (Lausanne) 2018, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Guise, T.; Kang, Y. The Biology of Bone Metastasis. Cold Spring Harb. Perspect. Med. 2018, 8, a031252. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Lester, J.E.; Dodwell, D.; Purohit, O.P.; Gutcher, S.A.; Ellis, S.P.; Thorpe, R.; Horsman, J.M.; Coleman, R.E. Prevention of anastrozole-induced bone loss with monthly oral ibandronate during adjuvant aromatase inhibitor therapy for breast cancer. Clin. Cancer Res. 2008, 14, 6336–6342. [Google Scholar] [CrossRef]
- von Moos, R.; Costa, L.; Gonzalez-Suarez, E.; Terpos, E.; Niepel, D.; Body, J.J. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat. Rev. 2019, 76, 57–67. [Google Scholar] [CrossRef]
- Yasuda, Y.; Li, Z.; Greenbaum, D.; Bogyo, M.; Weber, E.; Brömme, D. Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem. 2004, 279, 36761–36770. [Google Scholar] [CrossRef]
- Munari, E.; Cima, L.; Massari, F.; Bertoldo, F.; Porcaro, A.B.; Caliò, A.; Riva, G.; Ciocchetta, E.; Ciccarese, C.; Modena, A.; et al. Cathepsin K expression in castration-resistant prostate carcinoma: A therapeutical target for patients at risk for bone metastases. Int. J. Biol. Markers 2017, 32, e243–e247. [Google Scholar] [CrossRef]
- Jensen, A.B.; Wynne, C.; Ramirez, G.; He, W.; Song, Y.; Berd, Y.; Wang, H.; Mehta, A.; Lombardi, A. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: Results of a 4-week, double-blind, randomized, controlled trial. Clin. Breast Cancer 2010, 10, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Merck &Co. drops osteoporosis drug odanacatib. Nat. Rev. Drug Discov. 2016, 15, 669. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lee, Y.C.; Yu, G.; Cheng, C.J.; Zhou, X.; Chu, K.; Murshed, M.; Le, N.T.; Baseler, L.; Abe, J.I.; et al. Endothelial-to-osteoblast conversion generates osteoblastic metastasis of prostate cancer. Dev. Cell 2017, 41, 467–480. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis. Oncol. 2018, 2, 4. [Google Scholar] [CrossRef]
- Lynch, C.C. Matrix metalloproteinases as master regulators of the vicious cycle of bone metastasis. Bone 2011, 48, 44–53. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ECM Component | Proteinase |
|---|---|
| Versican (VCAN) | ADAMTs |
| Collagen I | MMP1, MMP3, MMP8, MMP10, MMP11, MMP13, MMP14, MMP19 ADAM8 |
| Collagen III | MMP1, MMP3, MMP8, MMP9, MMP10, MMP11, MMP13, MMP14, MMP16, |
| Collagen IV | MMP1, MMP2, MMP3, MMP7, MMP8, MMP9, MMP10, MMP11, MMP12, MMP13, MMP14, MMP19, MMP25, MMP26, ADAM15 Meprins |
| Fibronectin | MMP2, MMP7, MMP8, MMP9, MMP10, MMP11, MMP12, MMP13, MMP14, MMP15, MMP16, MMP19, MMP24, MMP25, MMP26 ADAM8, ADAM9, ADAM12, ADAMTs Meprins |
| TGF-β | Plasmin, MMP2, MMP9 |
| Periostin | ADAM8 |
| ECM Component | PMN Site | Mechanism | Reference |
|---|---|---|---|
| Periostin (POSTN) | Lung | Induces EMT, promotes lung metastases | [54] |
| Versican (VCAN) | Lung | Secreted by the infiltrating bone marrow-derived myeloid cells, promotes metastatic niche formation | [55] |
| Tenascin-C (TNC) | Lung | Secreted by breast cancer cells, promotes PMN formation | [56,57] |
| Vascular cell adhesion molecule-1 (VCAM-1) | Lung | Binds to α4β1 integrin, promotes homing of breast and melanoma cancer cells | [60,61,62] |
| Collagen I, III, IV | Lung, liver, bone | Increased collagen hydroxylation promotes ECM remodeling; increased collagen I turnover in hepatic metastases | [64,72,73,84,88] |
| MMP-1, MMP-2, MMP-9 | Lung | Increased MMPs expression promotes PMN formation | [65,66,67,68] |
| Fibronectin | Liver | Increased production in liver PMN; facilitates CTC extravasation | [74,75,76] |
| Atypical Cadherin 1 (FAT1) | Liver | Inactivation in liver metastases | [77] |
| Wnt/β-catenin axis | Liver | Aberrant signaling in liver metastases | [79,80,81] |
| CXCR-4/CXCL-12, CXCR-6/CXCL-16, CXCR-3/CXCL-10, | Bone | Promote bone metastases | [90] |
| TGF-β, PDGF, FGF, PTH-rP, BMPs Cathepsin K | Bone | Released during ECM degradation, promote bone metastases | [91,92,93,94] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolillo, M.; Schinelli, S. Extracellular Matrix Alterations in Metastatic Processes. Int. J. Mol. Sci. 2019, 20, 4947. https://doi.org/10.3390/ijms20194947
Paolillo M, Schinelli S. Extracellular Matrix Alterations in Metastatic Processes. International Journal of Molecular Sciences. 2019; 20(19):4947. https://doi.org/10.3390/ijms20194947
Chicago/Turabian StylePaolillo, Mayra, and Sergio Schinelli. 2019. "Extracellular Matrix Alterations in Metastatic Processes" International Journal of Molecular Sciences 20, no. 19: 4947. https://doi.org/10.3390/ijms20194947
APA StylePaolillo, M., & Schinelli, S. (2019). Extracellular Matrix Alterations in Metastatic Processes. International Journal of Molecular Sciences, 20(19), 4947. https://doi.org/10.3390/ijms20194947