Novel SCN5A p.W697X Nonsense Mutation Segregation in a Family with Brugada Syndrome


 , ,
, ,  ,
, 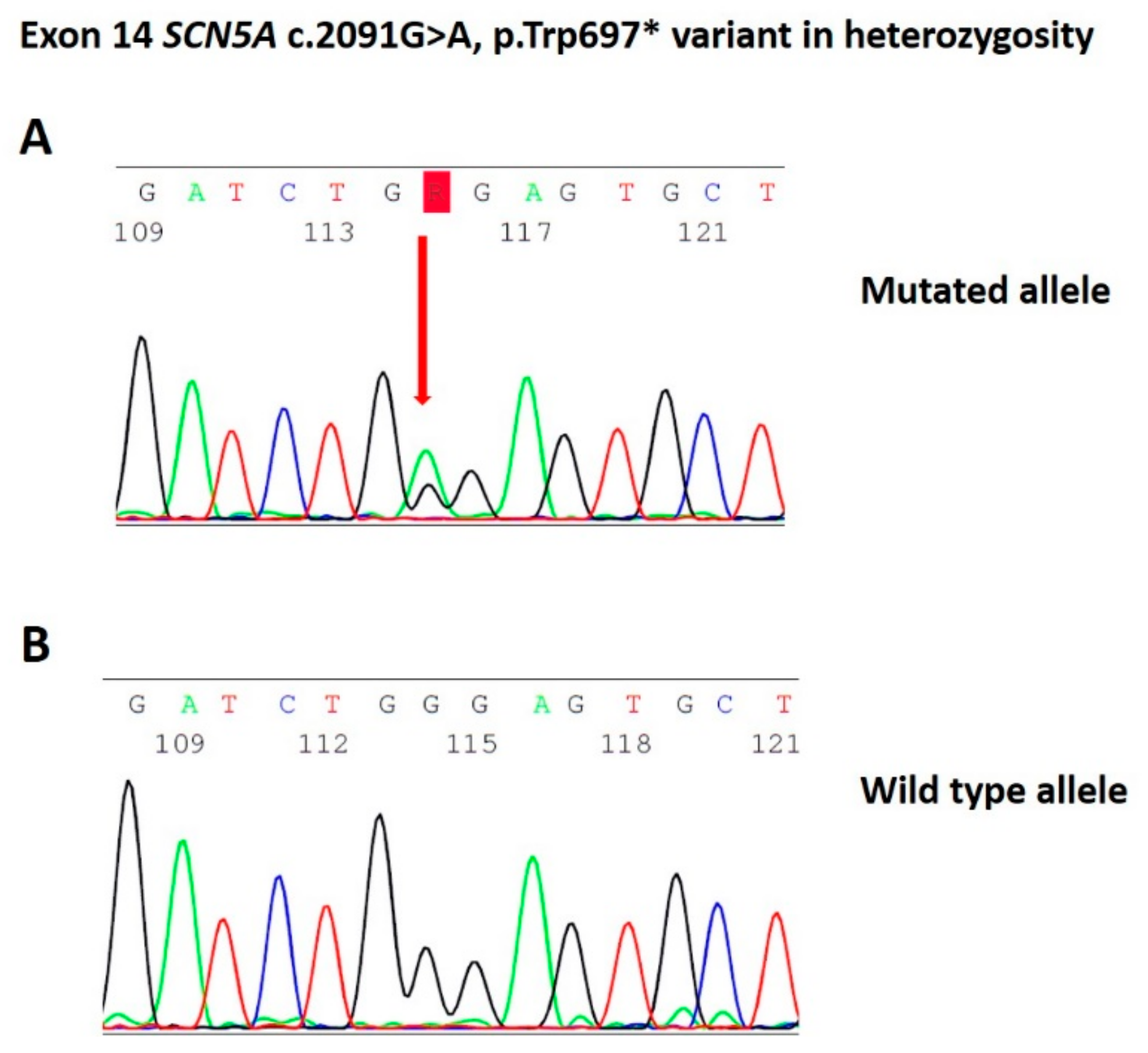{kind=link}
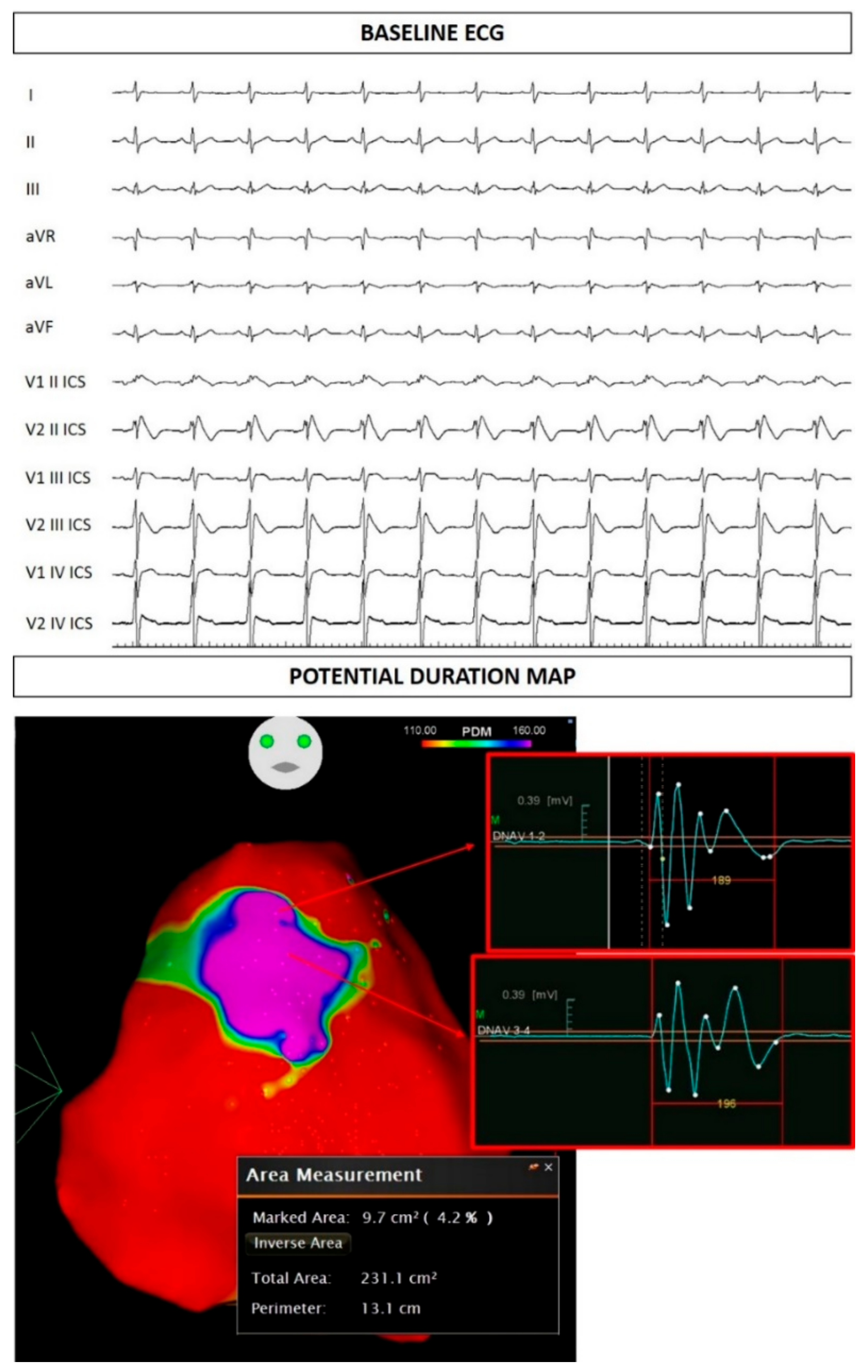{kind=link}
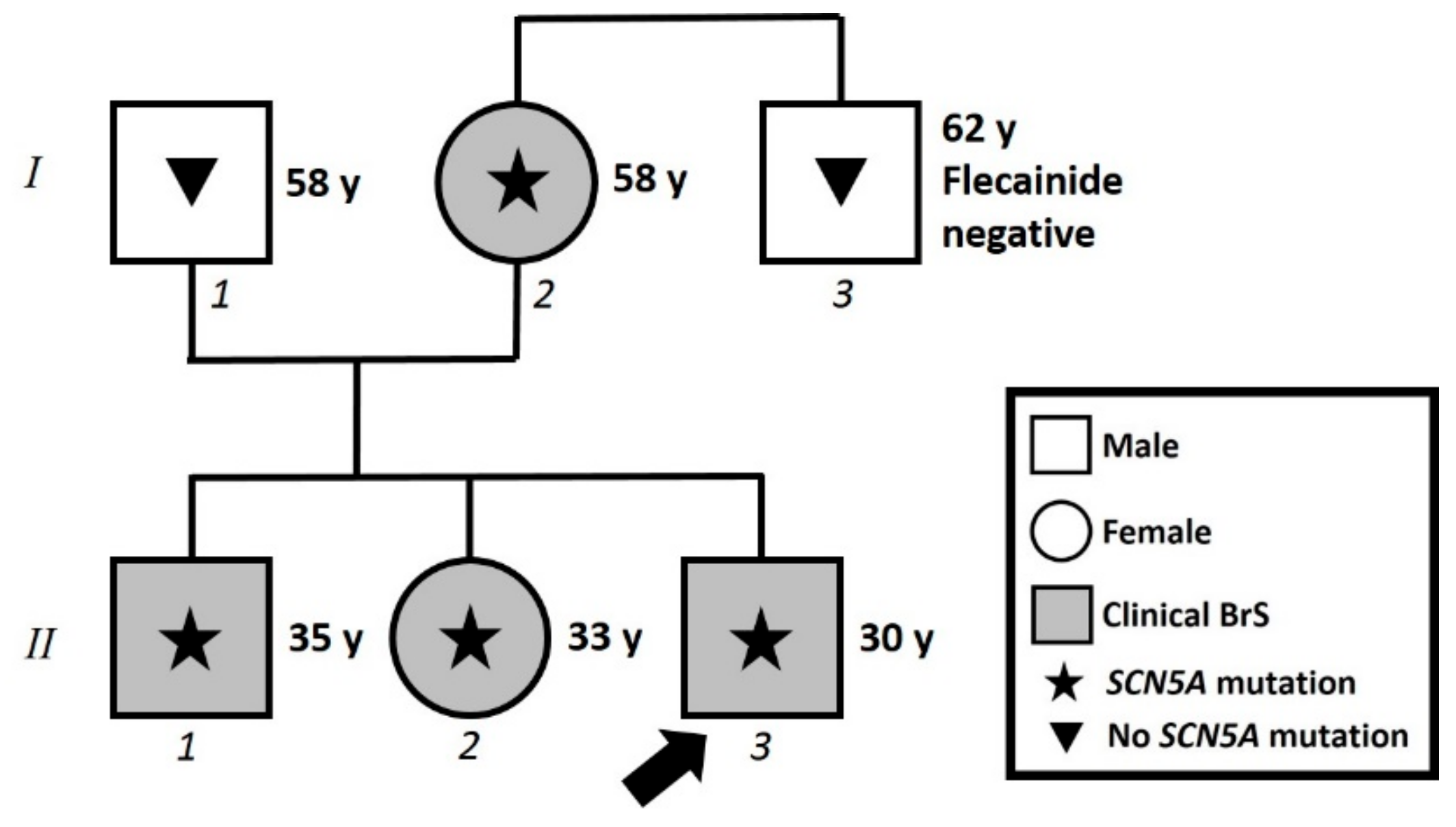{kind=link}
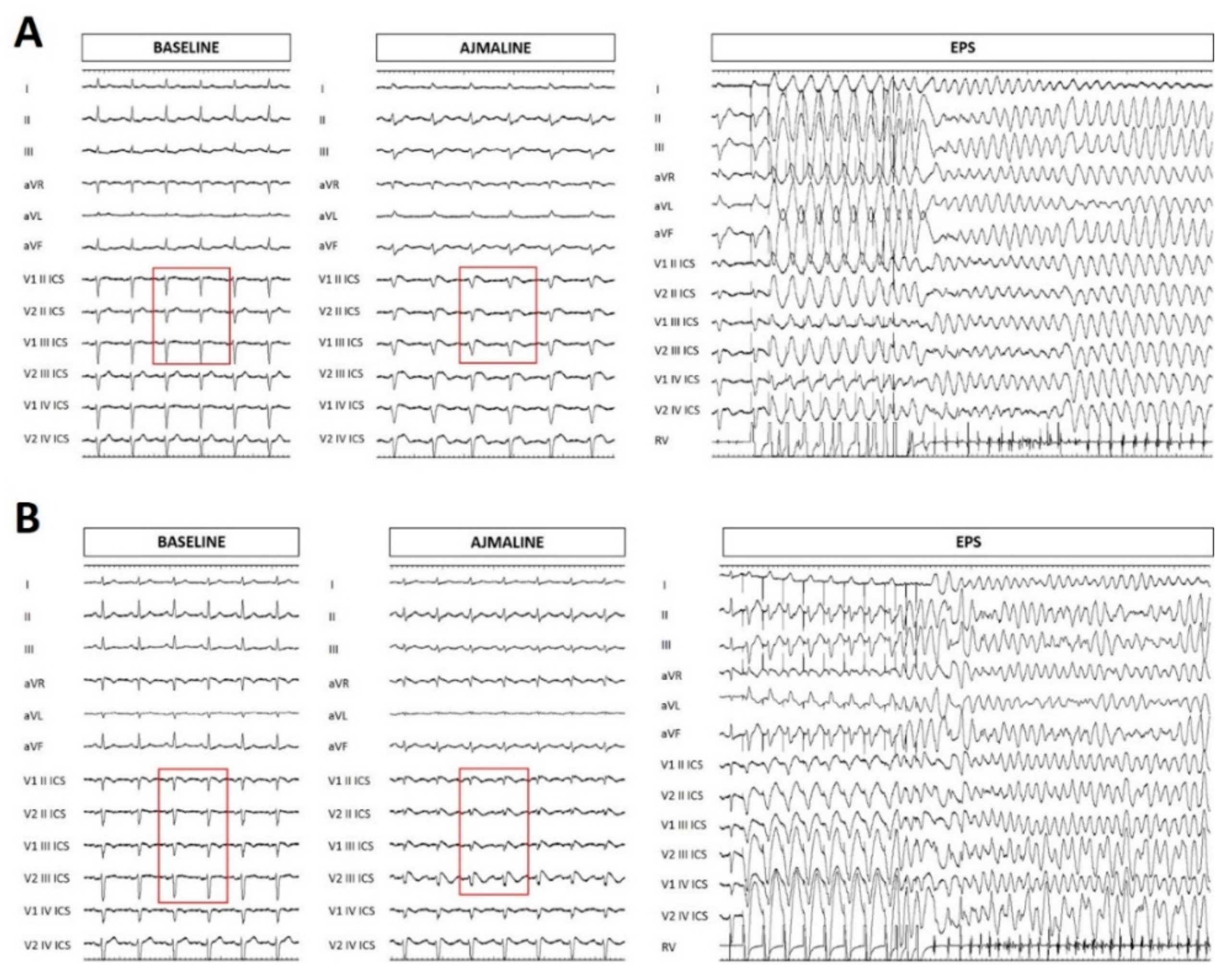{kind=link}
Abstract
1. Introduction
2. Results
2.1. Case Presentation
2.2. Assessment of Family Members
2.3. In Silico Predictions
3. Discussion
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Heart Rhythm 2016, 13, 295–324. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Pappone, C.; Piccoli, M.; Ghiroldi, A.; Micaglio, E.; Anastasia, L. Calcium in brugada syndrome: Questions for future research. Front Physiol. 2018, 9, 1088. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; Wilde, A.A. Inherited ion channel diseases: A brief review. Europace 2015, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Janin, A.; Bessiere, F.; Georgescu, T.; Chanavat, V.; Chevalier, P.; Millat, G. TRPM4 mutations to cause autosomal recessive and not autosomal dominant Brugada type 1 syndrome. Eur. J. Med. Genet. 2019, 62, 103527. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Zankov, D.P.; Ding, W.G.; Itoh, H.; Makiyama, T.; Doi, T.; Shizuta, S.; Hattori, T.; Miyamoto, A.; Naiki, N.; et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 2011, 4, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Tafti, M.F.; Khatami, M.; Rezaei, S.; Heidari, M.M.; Hadadzadeh, M. Novel and heteroplasmic mutations in mitochondrial tRNA genes in Brugada syndrome. Cardiol. J. 2018, 25, 113–119. [Google Scholar] [CrossRef]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Vicedomini, G.; Locati, E.T.; Ciconte, G.; Giannelli, L.; Giordano, F.; Crisa, S.; Vecchi, M.; Borrelli, V.; et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace 2019. [Google Scholar] [CrossRef]
- Pappone, C.; Monasky, M.; Ciconte, G. Epicardial ablation in genetic cardiomyopathies: A new frontier. Eur. Heart J. Suppl. 2019, 21, B61–B66. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Ciconte, G.; Anastasia, L.; Pappone, C. Commentary: next generation sequencing and linkage analysis for the molecular diagnosis of a novel overlapping syndrome characterized by hypertrophic cardiomyopathy and typical electrical instability of brugada syndrome. Front Physiol. 2017, 8, 1056. [Google Scholar] [CrossRef] [PubMed]
- Di Resta, C.; Pietrelli, A.; Sala, S.; Della Bella, P.; De Bellis, G.; Ferrari, M.; Bordoni, R.; Benedetti, S. High-throughput genetic characterization of a cohort of Brugada syndrome patients. Hum. Mol. Genet. 2015, 24, 5828–5835. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.; Santarpia, G.; Indolfi, C. The Brugada Syndrome- From Gene to Therapy. Circ. J. 2017, 81, 290–297. [Google Scholar] [CrossRef]
- Sieira, J.; Dendramis, G.; Brugada, P. Pathogenesis and management of Brugada syndrome. Nat. Rev. Cardiol. 2016, 13, 744–756. [Google Scholar] [CrossRef]
- Gosselin-Badaroudine, P.; Moreau, A.; Chahine, M. Nav 1.5 mutations linked to dilated cardiomyopathy phenotypes: Is the gating pore current the missing link? Channels 2014, 8, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zaklyazminskaya, E.; Dzemeshkevich, S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim. Biophys. Acta 2016, 1863, 1799–1805. [Google Scholar] [CrossRef]
- Micaglio, E.; Monasky, M.M.; Ciconte, G.; Vicedomini, G.; Conti, M.; Mecarocci, V.; Giannelli, L.; Giordano, F.; Pollina, A.; Saviano, M.; et al. SCN5A Nonsense Mutation and NF1 Frameshift Mutation in a Family With Brugada Syndrome and Neurofibromatosis. Front Genet. 2019, 10, 50. [Google Scholar] [CrossRef]
- Yeates, L.; Ingles, J.; Gray, B.; Singarayar, S.; Sy, R.W.; Semsarian, C.; Bagnall, R.D. A balanced translocation disrupting SCN5A in a family with Brugada syndrome and sudden cardiac death. Heart Rhythm 2019, 16, 231–238. [Google Scholar] [CrossRef]
- Yagihara, N.; Watanabe, H.; Barnett, P.; Duboscq-Bidot, L.; Thomas, A.C.; Yang, P.; Ohno, S.; Hasegawa, K.; Kuwano, R.; Chatel, S.; et al. Variants in the SCN5A Promoter Associated With Various Arrhythmia Phenotypes. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Micaglio, E.; Monasky, M.M.; Ciconte, G.; Vicedomini, G.; Conti, M.; Mecarocci, V.; Giannelli, L.; Giordano, F.; Pollina, A.; Saviano, M.; et al. Novel SCN5A Frameshift Mutation in Brugada Syndrome Associated With Complex Arrhythmic Phenotype. Front Genet. 2019, 10, 547. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Benedetti, S.; Di Resta, C.; Vicedomini, G.; Borrelli, V.; Ghiroldi, A.; Piccoli, M.; Anastasia, L.; et al. Genotype/Phenotype Relationship in a Consanguineal Family With Brugada Syndrome Harboring the R1632C Missense Variant in the SCN5A Gene. Front Physiol. 2019, 10, 666. [Google Scholar] [CrossRef]
- Sonoda, K.; Ohno, S.; Ozawa, J.; Hayano, M.; Hattori, T.; Kobori, A.; Yahata, M.; Aburadani, I.; Watanabe, S.; Matsumoto, Y.; et al. Copy Number Variations of SCN5A in Brugada Syndrome. SCN5A CNVs in BrS. Heart Rhythm 2018, 15, 1155–1188. [Google Scholar] [CrossRef] [PubMed]
- Ciconte, G.; Monasky, M.M.; Vicedomini, G.; Borrelli, V.; Giannelli, L.; Pappone, C. Unusual response to ajmaline test in Brugada syndrome patient leads to extracorporeal membrane oxygenator support. Europace 2019. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Brugada, P.; Brugada, R. The ajmaline challenge in Brugada syndrome: A useful tool or misleading information? Eur. Heart J. 2003, 24, 1085–1086. [Google Scholar] [CrossRef]
- VarSome: The Human Genomics Community. Available online: https://varsome.com/ (accessed on 4 July 2019).
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome. Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Gasparini, M.; Napolitano, C.; Della Bella, P.; Ottonelli, A.G.; Sassone, B.; Giordano, U.; Pappone, C.; Mascioli, G.; Rossetti, G.; et al. Risk stratification in Brugada syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J. Am. Coll. Cardiol. 2012, 59, 37–45. [Google Scholar] [CrossRef]
- Ciconte, G.; Santinelli, V.; Brugada, J.; Vicedomini, G.; Conti, M.; Monasky, M.M.; Borrelli, V.; Castracane, W.; Aloisio, T.; Giannelli, L.; et al. General Anesthesia Attenuates Brugada Syndrome Phenotype Expression: Clinical Implications From a Prospective Clinical Trial. JACC Clin. Electrophysiol. 2018, 4, 518–530. [Google Scholar] [CrossRef]
- Pappone, C.; Brugada, J.; Vicedomini, G.; Ciconte, G.; Manguso, F.; Saviano, M.; Vitale, R.; Cuko, A.; Giannelli, L.; Calovic, Z.; et al. Electrical Substrate Elimination in 135 Consecutive Patients With Brugada Syndrome. Circ. Arrhythm. Electrophysiol. 2017, 10, e005053. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Ciconte, G.; Manguso, F.; Vicedomini, G.; Mecarocci, V.; Conti, M.; Giannelli, L.; Pozzi, P.; Borrelli, V.; Menicanti, L.; et al. Assessing the Malignant Ventricular Arrhythmic Substrate in Patients With Brugada Syndrome. J. Am. Coll. Cardiol. 2018, 71, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Sroubek, J.; Probst, V.; Mazzanti, A.; Delise, P.; Hevia, J.C.; Ohkubo, K.; Zorzi, A.; Champagne, J.; Kostopoulou, A.; Yin, X.; et al. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation 2016, 133, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Herfst, L.J.; Potet, F.; Bezzina, C.R.; Groenewegen, W.A.; Le Marec, H.; Hoorntje, T.M.; Demolombe, S.; Baro, I.; Escande, D.; Jongsma, H.J.; et al. Na+ channel mutation leading to loss of function and non-progressive cardiac conduction defects. J. Mol. Cell. Cardiol. 2003, 35, 549–557. [Google Scholar] [CrossRef]
- Tfelt-Hansen, J.; Jespersen, T.; Hofman-Bang, J.; Rasmussen, H.B.; Cedergreen, P.; Skovby, F.; Abriel, H.; Svendsen, J.H.; Olesen, S.P.; Christiansen, M.; et al. Ventricular tachycardia in a Brugada syndrome patient caused by a novel deletion in SCN5A. Can. J. Cardiol. 2009, 25, 156–160. [Google Scholar] [CrossRef][Green Version]
- Valdivia, C.R.; Tester, D.J.; Rok, B.A.; Porter, C.B.; Munger, T.M.; Jahangir, A.; Makielski, J.C.; Ackerman, M.J. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 2004, 62, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, F.; Probst, V.; Potet, F.; Demolombe, S.; Chevallier, J.C.; Baro, I.; Moisan, J.P.; Boisseau, P.; Schott, J.J.; Escande, D.; et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 2001, 104, 3081–3086. [Google Scholar] [CrossRef]
- Bezzina, C.; Veldkamp, M.W.; Van Den Berg, M.P.; Postma, A.V.; Rook, M.B.; Viersma, J.W.; van Langen, I.M.; Tan-Sindhunata, G.; Bink-Boelkens, M.T.; Van Der Hout, A.H.; et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 1999, 85, 1206–1213. [Google Scholar] [CrossRef]
- Dumaine, R.; Towbin, J.A.; Brugada, P.; Vatta, M.; Nesterenko, D.V.; Nesterenko, V.V.; Brugada, J.; Brugada, R.; Antzelevitch, C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ. Res. 1999, 85, 803–809. [Google Scholar] [CrossRef]
- Maury, P.; Moreau, A.; Hidden-Lucet, F.; Leenhardt, A.; Fressart, V.; Berthet, M.; Denjoy, I.; Bennamar, N.; Rollin, A.; Cardin, C.; et al. Novel SCN5A mutations in two families with “Brugada-like” ST elevation in the inferior leads and conduction disturbances. J. Interv. Card. Electrophysiol. 2013, 37, 131–140. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micaglio, E.; Monasky, M.M.; Resta, N.; Bagnulo, R.; Ciconte, G.; Giannelli, L.; Locati, E.T.; Vicedomini, G.; Borrelli, V.; Ghiroldi, A.; et al. Novel SCN5A p.W697X Nonsense Mutation Segregation in a Family with Brugada Syndrome. Int. J. Mol. Sci. 2019, 20, 4920. https://doi.org/10.3390/ijms20194920
Micaglio E, Monasky MM, Resta N, Bagnulo R, Ciconte G, Giannelli L, Locati ET, Vicedomini G, Borrelli V, Ghiroldi A, et al. Novel SCN5A p.W697X Nonsense Mutation Segregation in a Family with Brugada Syndrome. International Journal of Molecular Sciences. 2019; 20(19):4920. https://doi.org/10.3390/ijms20194920
Chicago/Turabian StyleMicaglio, Emanuele, Michelle M. Monasky, Nicoletta Resta, Rosanna Bagnulo, Giuseppe Ciconte, Luigi Giannelli, Emanuela T. Locati, Gabriele Vicedomini, Valeria Borrelli, Andrea Ghiroldi, and et al. 2019. "Novel SCN5A p.W697X Nonsense Mutation Segregation in a Family with Brugada Syndrome" International Journal of Molecular Sciences 20, no. 19: 4920. https://doi.org/10.3390/ijms20194920
APA StyleMicaglio, E., Monasky, M. M., Resta, N., Bagnulo, R., Ciconte, G., Giannelli, L., Locati, E. T., Vicedomini, G., Borrelli, V., Ghiroldi, A., Anastasia, L., Benedetti, S., Di Resta, C., Ferrari, M., & Pappone, C. (2019). Novel SCN5A p.W697X Nonsense Mutation Segregation in a Family with Brugada Syndrome. International Journal of Molecular Sciences, 20(19), 4920. https://doi.org/10.3390/ijms20194920