Novel Genetic Markers for Early Detection of Elevated Breast Cancer Risk in Women



Abstract
1. Introduction
2. Results and Discussion
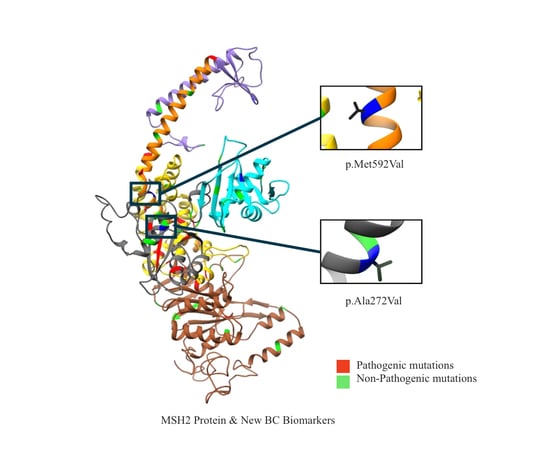
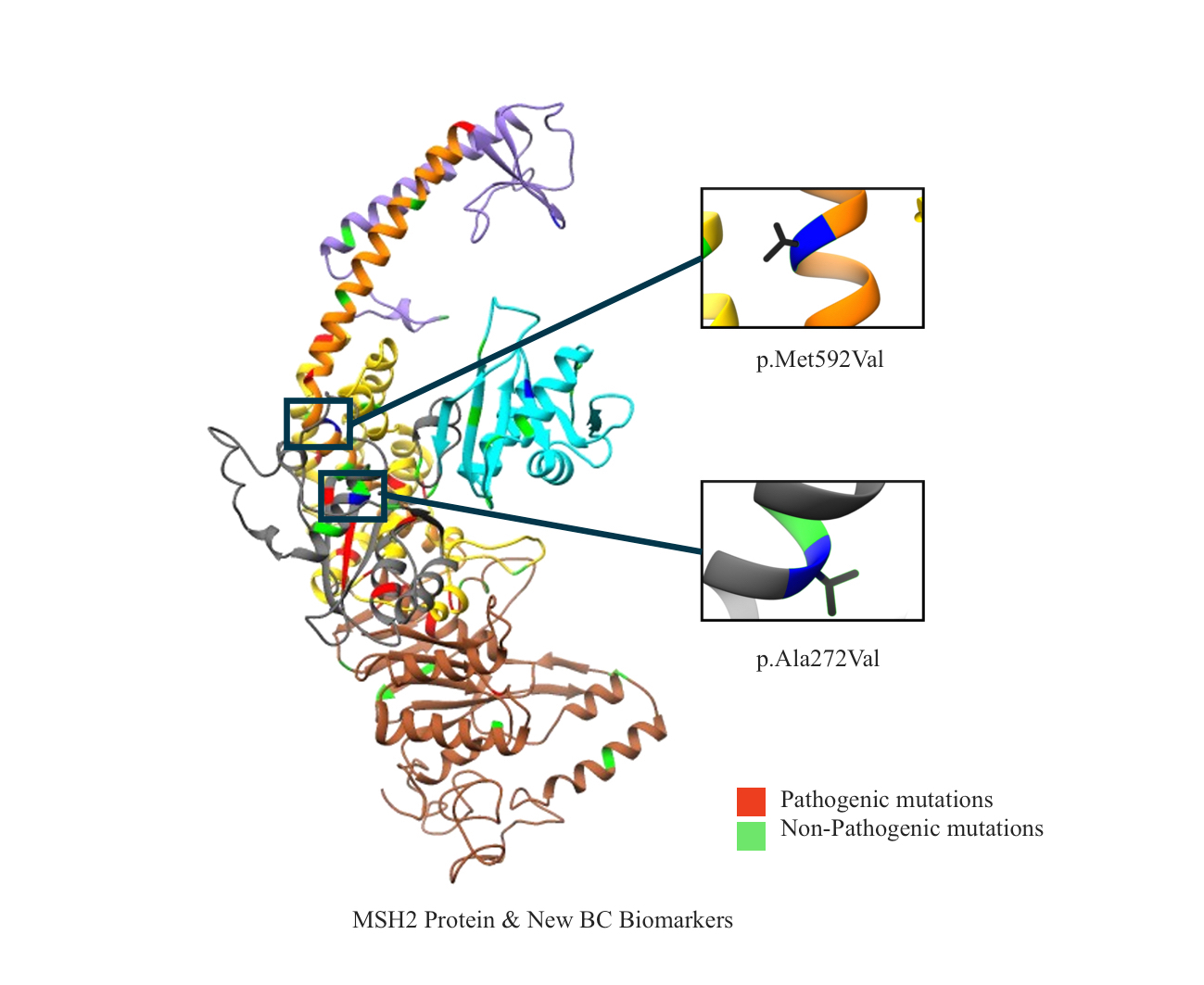
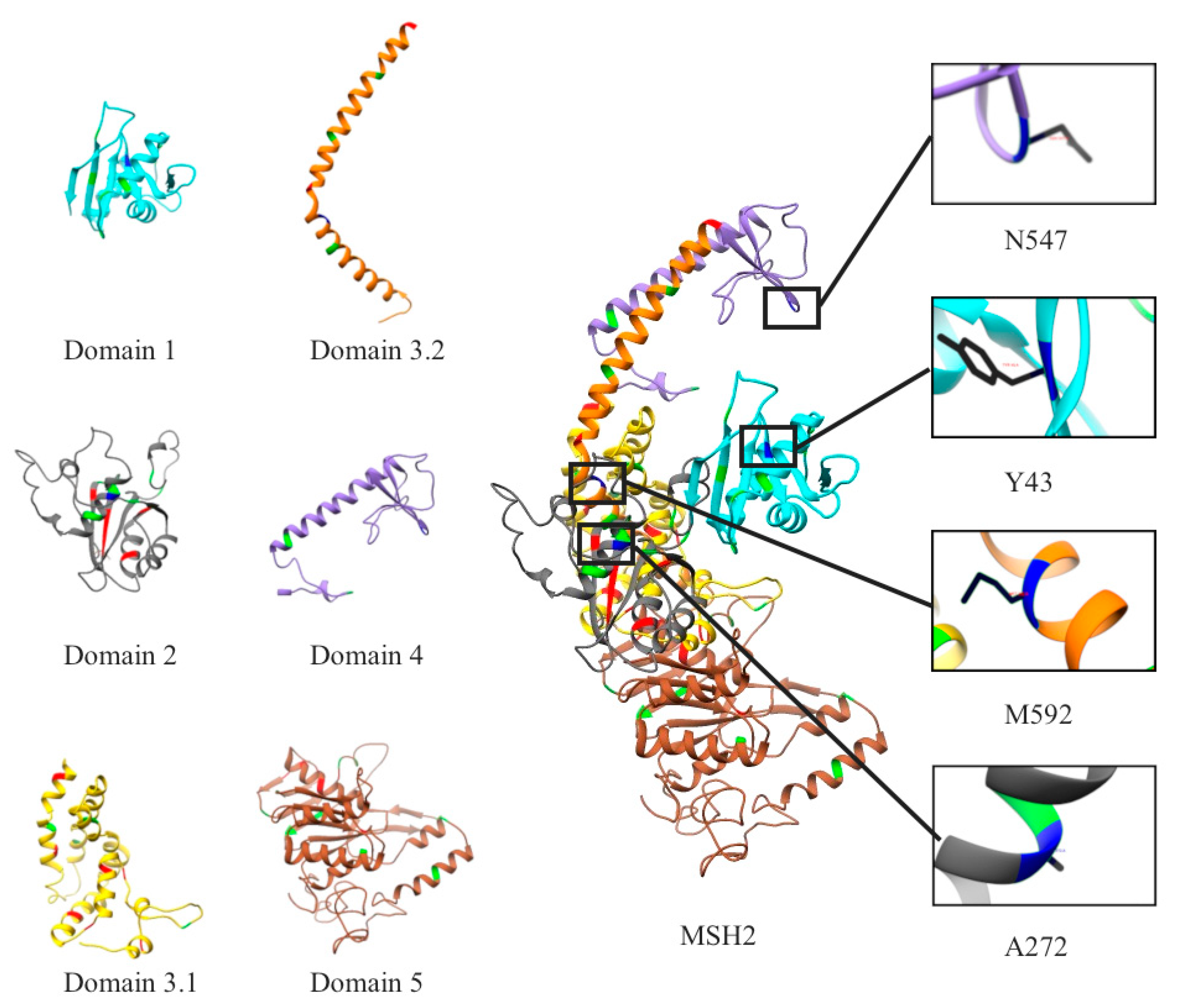
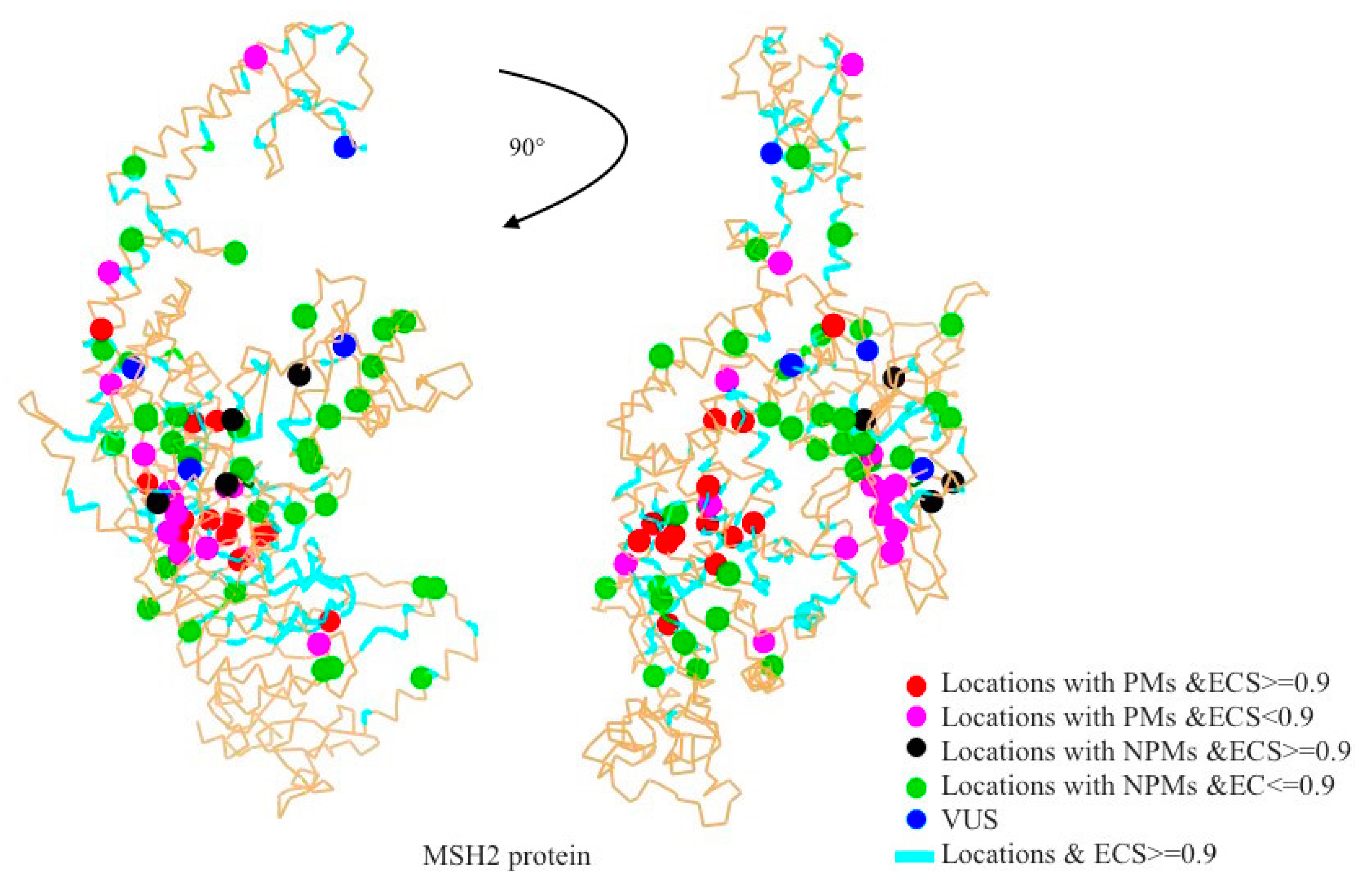
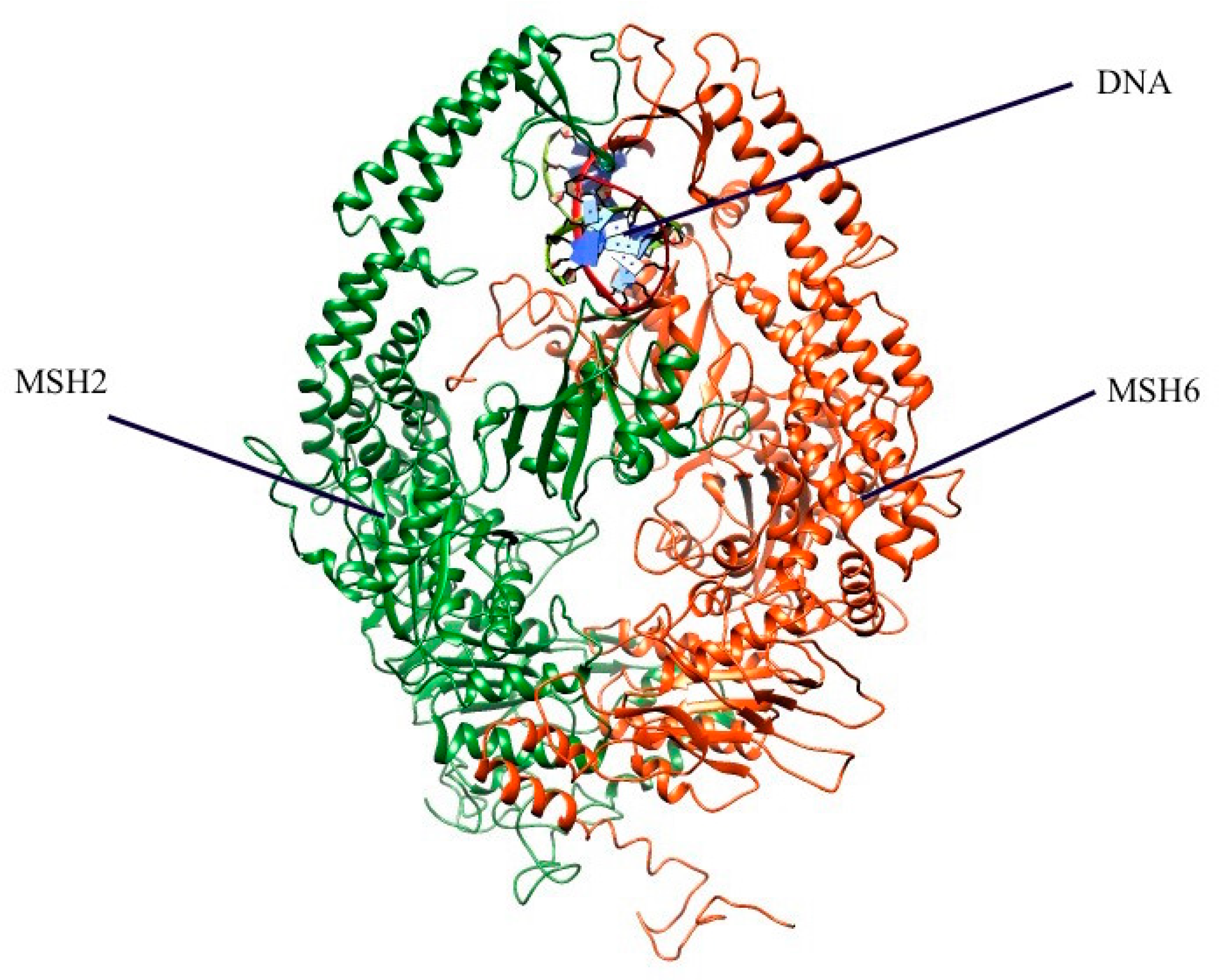
2.1. Mapping All Missense Mutations onto the 3D Structure of MSH2 Protein
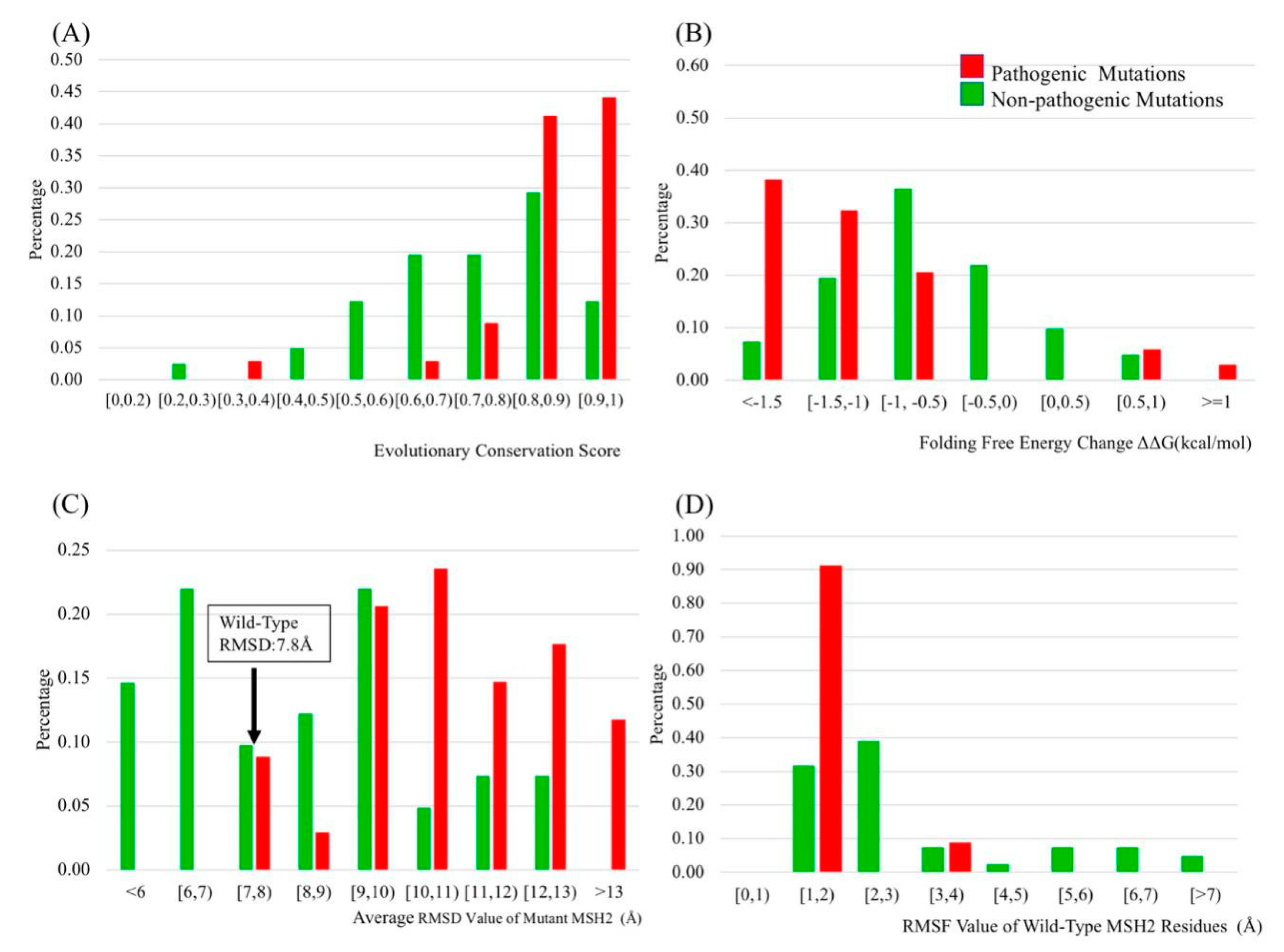
2.2. Evolutionary Conservation Score (ECS)
2.3. Folding and Binding Free Energy Changes
2.4. Molecular Dynamics (MD) Simulation
2.5. B-Factors
2.6. Relative Solvent Accessible Surface Area (rSASA) and B-Factors
2.7. Protein Distance (PD)
2.8. Hydrogen Bonds
2.9. Receiver Operating Characteristics (ROC)
2.10. Selecting the Best Predicting Protocol
2.11. Classification of VUS Using KNN Method
2.12. Clinical Features of VUS
3. Materials and Methods
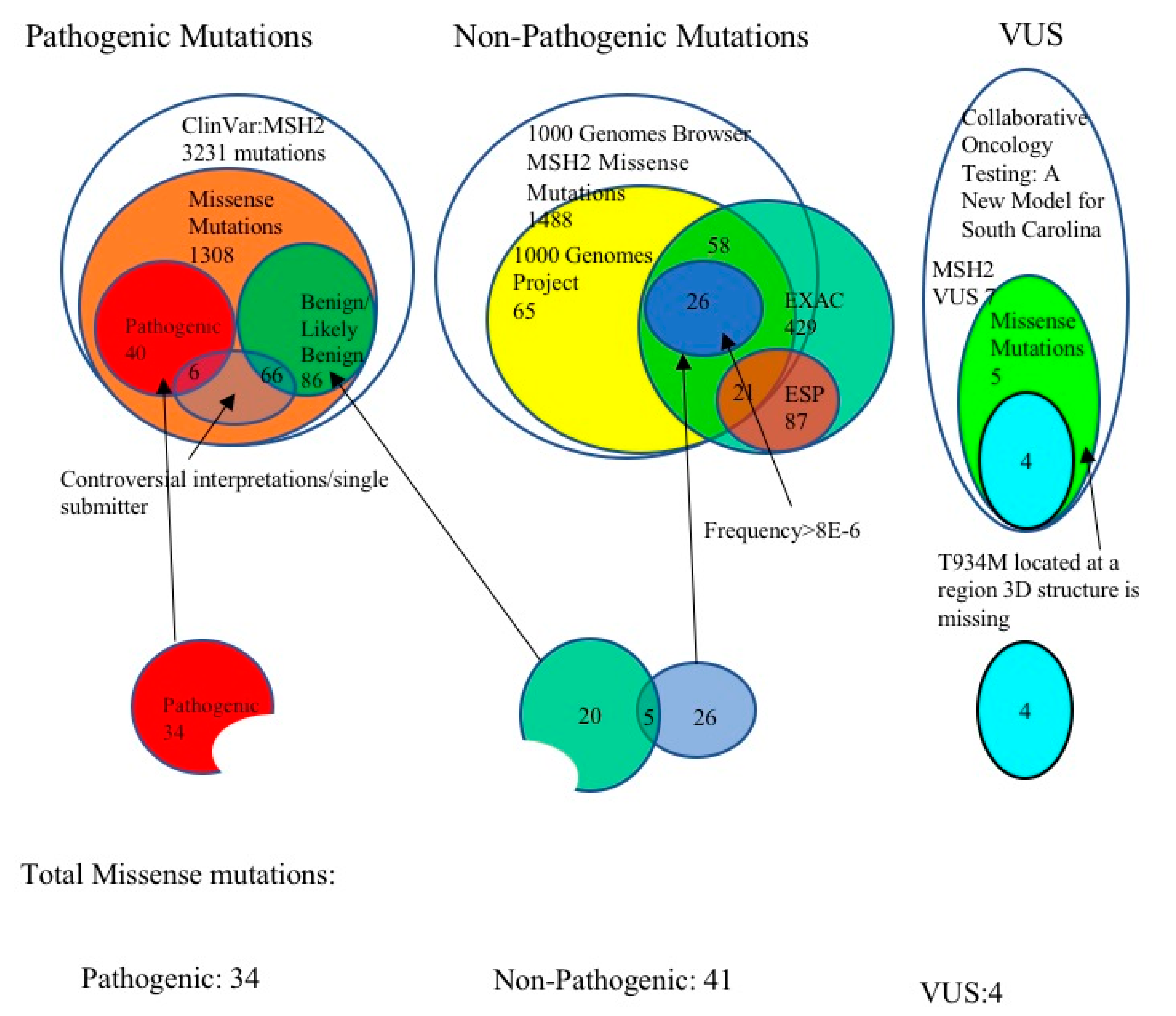
3.1. Selection of Pathogenic MSH2 Mutations
3.2. Selection of Benign MSH2 Mutations
3.3. MSH2 Missense Variants of Uncertain Significance
3.4. Preparation of 3D Structure of MSH2
3.5. Property Distance (PD)
3.6. Evolutionary Conservation Score (ECS) Calculations
3.7. Folding Free Energy Change (ΔΔG) and Binding Free Energy Change (ΔΔΔG)
3.8. Relative Solvent Accessible Surface Area (rSASA) and Hydrogen Bond (H-bond) Number Calculations
3.9. B-Factor of the Alpha Carbonate of the Corresponding Residue
3.10. Molecular Dynamic (MD) Simulations
3.11. K-Nearest Neighbors (KNN) and Support Vector Machine (SVM) Classifications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MMR | Mismatch Repair |
| LS | Lynch Syndrome |
| EXAC | Exome Aggregation Consortium |
| BC | Breast Cancer |
| AUC | Area Under the Curve |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuations |
| rSASA | Relative solvent accessible surface area |
| MD | Molecular Dynamics |
| PD | Protein Distance |
| KNN | K-nearest neighbors |
| SVM | Support Vector Machine |
| WT | Wild-Type |
| GI | Gastro-Intestinal Cancer |
| VUS | Variants of Unknown Significance |
| PMs | Pathogenic Mutations |
| NPMs | Non-Pathogenic Mutations |
| ESP | NHLBI-GO Exome Sequencing Project Exome Variant Server |
| ECS | Evolutionary Conservation Score |
References
- Marti, T.M.; Kunz, C.; Fleck, O. DNA mismatch repair and mutation avoidance pathways. J Cell. Physiol. 2002, 191, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Bowen, N.; Kolodner, R.D. Mispair-specific recruitment of the Mlh1-Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2-Msh3 complex. J. Biol. Chem. 2014, 289, 9352–9364. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Lynch, H.T.; Smyrk, T.; Lynch, J.; Fitzgibbons, R., Jr.; Lanspa, S.; McGinn, T. Update on the differential diagnosis, surveillance and management of hereditary non-polyposis colorectal cancer. Eur. J. Cancer 1995, 31, 1039–1046. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, M.W.; Magnuson, C.W.; Larsen, A.L.; Krush, A.J. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch. Intern. Med. 1966, 117, 206–212. [Google Scholar] [CrossRef]
- Lynch, H.T.; Krush, A.J.; Larsen, A.L. Heredity and endometrial carcinoma. South. Med. J. 1967, 60, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Krush, A.J. Heredity and adenocarcinoma of the colon. Gastroenterology 1967, 53, 517–527. [Google Scholar] [CrossRef]
- Parc, Y.; Boisson, C.; Thomas, G.; Olschwang, S. Cancer risk in 348 French MSH2 or MLH1 gene carriers. J. Med. Genet. 2003, 40, 208–213. [Google Scholar] [CrossRef]
- Barrow, E.; Alduaij, W.; Robinson, L.; Shenton, A.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Colorectal cancer in HNPCC: Cumulative lifetime incidence, survival and tumour distribution. A report of 121 families with proven mutations. Clin. Genet. 2008, 74, 233–242. [Google Scholar] [CrossRef]
- Lynch, H.T.; Coronel, S.M.; Okimoto, R.; Hampel, H.; Sweet, K.; Lynch, J.F.; Barrows, A.; Wijnen, J.; van der Klift, H.; Franken, P.; et al. A founder mutation of the MSH2 gene and hereditary nonpolyposis colorectal cancer in the United States. JAMA 2004, 291, 718–724. [Google Scholar] [CrossRef]
- Janavicius, R.; Elsakov, P. Novel germline MSH2 mutation in lynch syndrome patient surviving multiple cancers. Hered Cancer Clin. Pr. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Yamashita, K.; Yamashita, N.; Nakatsumi, Y.; Esumi, H.; Kawashima, A.; Ohta, T.; Mai, M.; Minamoto, T. Non-Hodgkin’s lymphoma in a patient with probable hereditary nonpolyposis colon cancer: Report of a case and review of the literature. Dis. Colon Rectum 2002, 45, 273–279. [Google Scholar] [CrossRef]
- Joost, P.; Therkildsen, C.; Dominguez-Valentin, M.; Jonsson, M.; Nilbert, M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology 2015, 86, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Bray, T.; Cannon, J.A.; Cooper, G.; David, D.S.; Early, D.S.; Erwin, D.; Ford, J.M.; et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1010–1030. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, D.; McLeod, R.; Graham, G.; Steckley, J.L.; Booth, K.; Somerville, M.J.; Andrew, S.E. A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple cafe-au-lait spots. Cancer Res. 2002, 62, 359–362. [Google Scholar] [PubMed]
- Zhu, M.; Chen, H.M.; Wang, Y.P. Missense mutations of MLH1 and MSH2 genes detected in patients with gastrointestinal cancer are associated with exonic splicing enhancers and silencers. Oncol. Lett. 2013, 5, 1710–1718. [Google Scholar] [CrossRef][Green Version]
- Takano, K.; Ichikawa, Y.; Ueno, E.; Ohwada, M.; Suzuki, M.; Tsunoda, H.; Miwa, M.; Uchida, K.; Yoshikawa, H. Microsatellite instability and expression of mismatch repair genes in sporadic endometrial cancer coexisting with colorectal or breast cancer. Oncol. Rep. 2005, 13, 11–16. [Google Scholar] [CrossRef]
- Lacroix-Triki, M.; Lambros, M.B.; Geyer, F.C.; Suarez, P.H.; Reis-Filho, J.S.; Weigelt, B. Absence of microsatellite instability in mucinous carcinomas of the breast. Int. J. Clin. Exp. Pathol. 2010, 4, 22–31. [Google Scholar]
- Goldberg, M.; Bell, K.; Aronson, M.; Semotiuk, K.; Pond, G.; Gallinger, S.; Zbuk, K. Association between the Lynch syndrome gene MSH2 and breast cancer susceptibility in a Canadian familial cancer registry. J. Med. Genet. 2017, 54, 742–746. [Google Scholar] [CrossRef]
- Rashid, M.U.; Naeemi, H.; Muhammad, N.; Loya, A.; Yusuf, M.A.; Lubinski, J.; Jakubowska, A.; Hamann, U. A novel deleterious c.2656G>T MSH2 germline mutation in a Pakistani family with a phenotypic overlap of hereditary breast and ovarian cancer and Lynch syndrome. Hered Cancer Clin. Pract. 2016, 14, 14. [Google Scholar] [CrossRef]
- Warren, J.J.; Pohlhaus, T.J.; Changela, A.; Iyer, R.R.; Modrich, P.L.; Beese, L.S. Structure of the human MutSalpha DNA lesion recognition complex. Mol. Cell 2007, 26, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Mazur, D.J.; Mendillo, M.L.; Kolodner, R.D. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol. Cell 2006, 22, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ollila, S.; Sarantaus, L.; Kariola, R.; Chan, P.; Hampel, H.; Holinski-Feder, E.; Macrae, F.; Kohonen-Corish, M.; Gerdes, A.M.; Peltomaki, P.; et al. Pathogenicity of MSH2 missense mutations is typically associated with impaired repair capability of the mutated protein. Gastroenterology 2006, 131, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Taverna, P.; Liu, L.; Hanson, A.J.; Monks, A.; Gerson, S.L. Characterization of MLH1 and MSH2 DNA mismatch repair proteins in cell lines of the NCI anticancer drug screen. Cancer Chemother. Pharmacol. 2000, 46, 507–516. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- ESP. NHLBI-GO Exome Sequencing Project Exome Variant Server; ESP (Exome Sequencing Project): Seattle, WA, USA, 2017. [Google Scholar]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Capriotti, E.; Altman, R.B. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinform. 2011, 12, S3. [Google Scholar] [CrossRef]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Getov, I.; Petukh, M.; Alexov, E. SAAFEC: Predicting the Effect of Single Point Mutations on Protein Folding Free Energy Using a Knowledge-Modified MM/PBSA Approach. Int. J. Mol. Sci. 2016, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- Topham, C.M.; Srinivasan, N.; Blundell, T.L. Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Prot. Eng. 1997, 10, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Simonetti, F.L.; Goncearenco, A.; Panchenko, A.R. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016, 44, W494–W501. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Dai, L.; Alexov, E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. Int. J. Mol. Sci. 2016, 17, 547. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef]
- Nilbert, M.; Wikman, F.P.; Hansen, T.V.; Krarup, H.B.; Orntoft, T.F.; Nielsen, F.C.; Sunde, L.; Gerdes, A.M.; Cruger, D.; Timshel, S.; et al. Major contribution from recurrent alterations and MSH6 mutations in the Danish Lynch syndrome population. Fam. Cancer 2009, 8, 75–83. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Evans, D.G.R.; Nakken, S.; Tubeuf, H.; Vodak, D.; Ekstrom, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Martins, A.; et al. Genetic variants of prospectively demonstrated phenocopies in BRCA1/2 kindreds. Hered Cancer Clin. Pract. 2018, 16, 4. [Google Scholar] [CrossRef]
- Borg, A.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Torngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat 2010, 31, E1200–E1240. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 14 November 2017).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed]
- Petrey, D.; Xiang, Z.; Tang, C.L.; Xie, L.; Gimpelev, M.; Mitros, T.; Soto, C.S.; Goldsmith-Fischman, S.; Kernytsky, A.; Schlessinger, A.; et al. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 2003, 53, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Myers, R.; Zhang, W.; Alexov, E. Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. Int. J. Mol. Sci 2018, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Petukh, M.; Li, M.; Alexov, E. Predicting Binding Free Energy Change Caused by Point Mutations with Knowledge-Modified MM/PBSA Method. PLoS Comput. Biol. 2015, 11, e1004276. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Sun, L.; Jia, Z.; Li, L.; Alexov, E. Predicting protein-DNA binding free energy change upon missense mutations using modified MM/PBSA approach: SAMPDI webserver. Bioinformatics 2018, 34, 779–786. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wu, B.; Eggert, J.; Alexov, E. Molecular mechanisms underlying pathogenic missense mutations. eLS 2014. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifiers | AUC Value | Classifiers | AUC Value |
|---|---|---|---|
| Folding free energy change | 0.77 | H-bond numbers of mutant residues | 0.54 |
| Binding free energy change | 0.53 | H-bond numbers of wild-type residues | 0.52 |
| Evolutionary conservation score (ECS) | 0.81 | H-bond number change upon mutations | 0.52 |
| Average root mean square deviations (RMSD) of all protein structures | 0.80 | B-factors of wild-type residues | 0.69 |
| RMSD change upon mutations | 0.65 | Relative solvent accessible surface area (rSASA) of mutation residues | 0.69 |
| Root mean square fluctuations (RMSF) of mutant residue | 0.69 | rSASA of wild-type residues | 0.72 |
| RMSF of wild-type residue | 0.80 | rSASA change upon mutations | 0.50 |
| RMSF change upon mutations | 0.73 | Protein Distance | 0.70 |
| Cumulative RMSF of mutant | 0.57 | Residue size change upon mutations | 0.70 |
| Cumulative RMSF of wild-type | 0.61 | Residue charge change upon mutations | 0.54 |
| Cumulative RMSF change upon mutation | 0.55 | Polarity change upon mutations | 0.70 |
| MSH2 VUS Mutations | p.Tyr43Cys | p.Ala272Val | p.Asn547Ser | p.Met592Val |
|---|---|---|---|---|
| Folding ∆∆G | −1.228 | −0.788 | −0.26 | −1.286 |
| ECS | 0.767 | 0.877 | 0.863 | 0.575 |
| RMSF_WT | 4.559 | 1.966 | 5.725 | 1.876 |
| Prediction | Non-pathogenic | Pathogenic | Non-pathogenic | Pathogenic |
| BC Mutations | BRCA2: c.4936_4939delGAAA | N/A | BRCA2: c.8791A>G | N/A |
| Clinical feature | Intermediate bilateral stage III size>1 | High grade stage I BC size<1cm | Intermediate, stage II A size<1cm | Intermediate stage II A |
| Family History of BC/GI | Yes/Yes | Yes/No | Yes/No | Yes/No |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Peng, Y.; Eggert, J.; Alexov, E. Novel Genetic Markers for Early Detection of Elevated Breast Cancer Risk in Women. Int. J. Mol. Sci. 2019, 20, 4828. https://doi.org/10.3390/ijms20194828
Wu B, Peng Y, Eggert J, Alexov E. Novel Genetic Markers for Early Detection of Elevated Breast Cancer Risk in Women. International Journal of Molecular Sciences. 2019; 20(19):4828. https://doi.org/10.3390/ijms20194828
Chicago/Turabian StyleWu, Bohua, Yunhui Peng, Julia Eggert, and Emil Alexov. 2019. "Novel Genetic Markers for Early Detection of Elevated Breast Cancer Risk in Women" International Journal of Molecular Sciences 20, no. 19: 4828. https://doi.org/10.3390/ijms20194828
APA StyleWu, B., Peng, Y., Eggert, J., & Alexov, E. (2019). Novel Genetic Markers for Early Detection of Elevated Breast Cancer Risk in Women. International Journal of Molecular Sciences, 20(19), 4828. https://doi.org/10.3390/ijms20194828