Vascular Endothelial Cell Biology: An Update




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Luminal Endothelial Cell Surface, the Glycocalyx
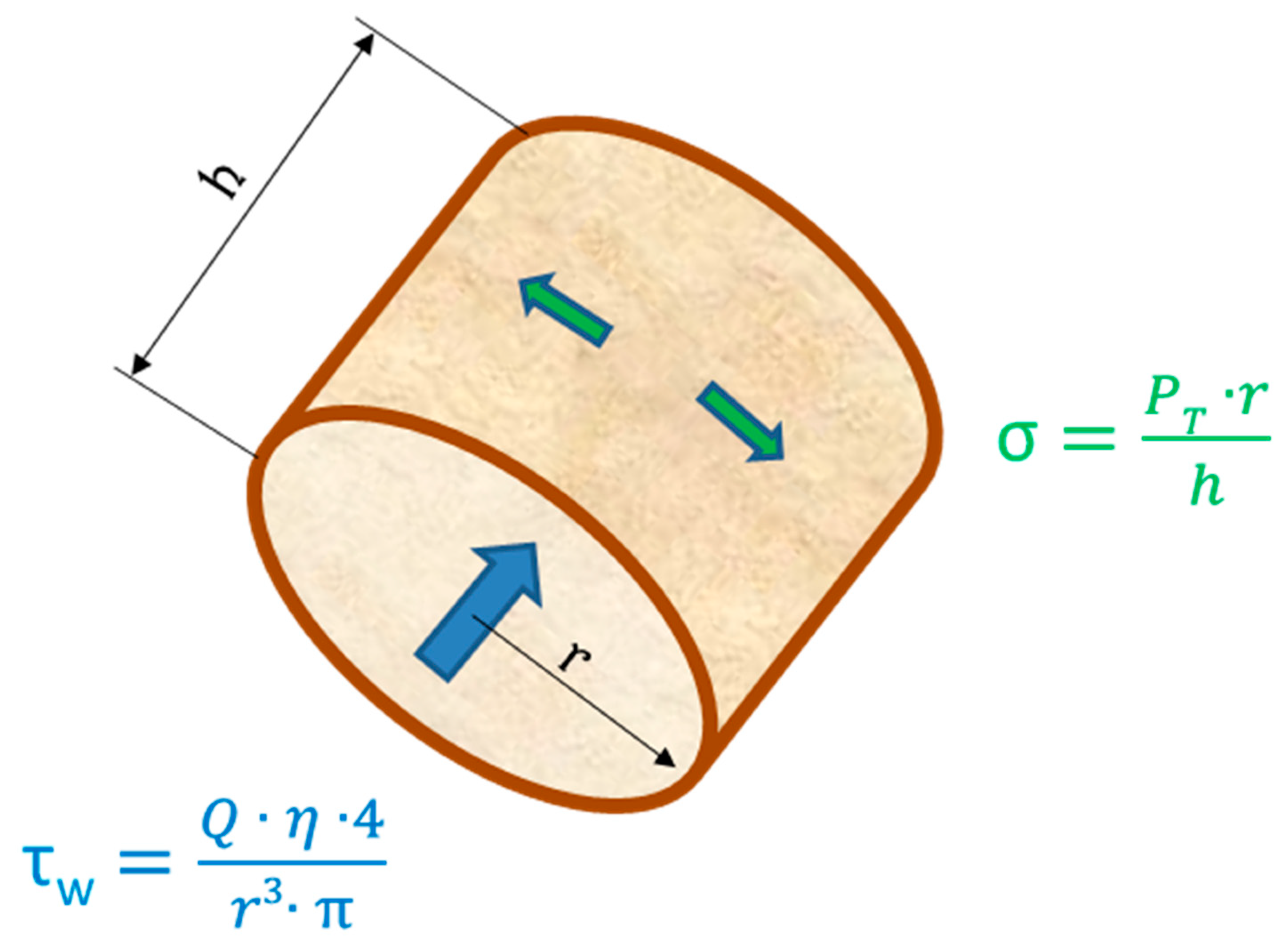
3. Vascular Endothelium and Shear Stress
4. Endothelial Cells and Blood Flow
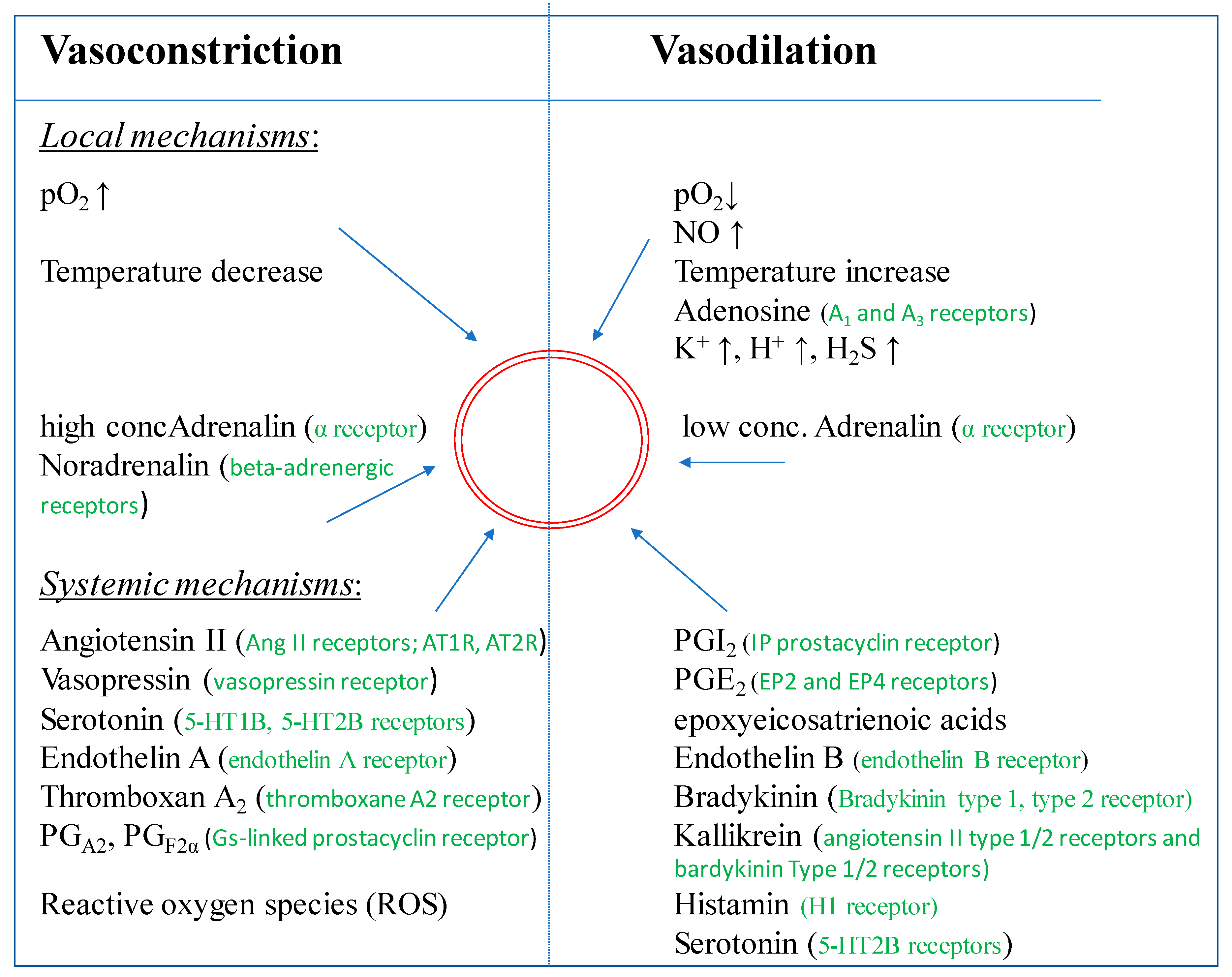
4.1. Regulation by Vasodilation
4.2. Regulation by Vasoconstriction
5. Endothelial Cells and Thrombosis
6. Heterogeneity of Endothelial Cells
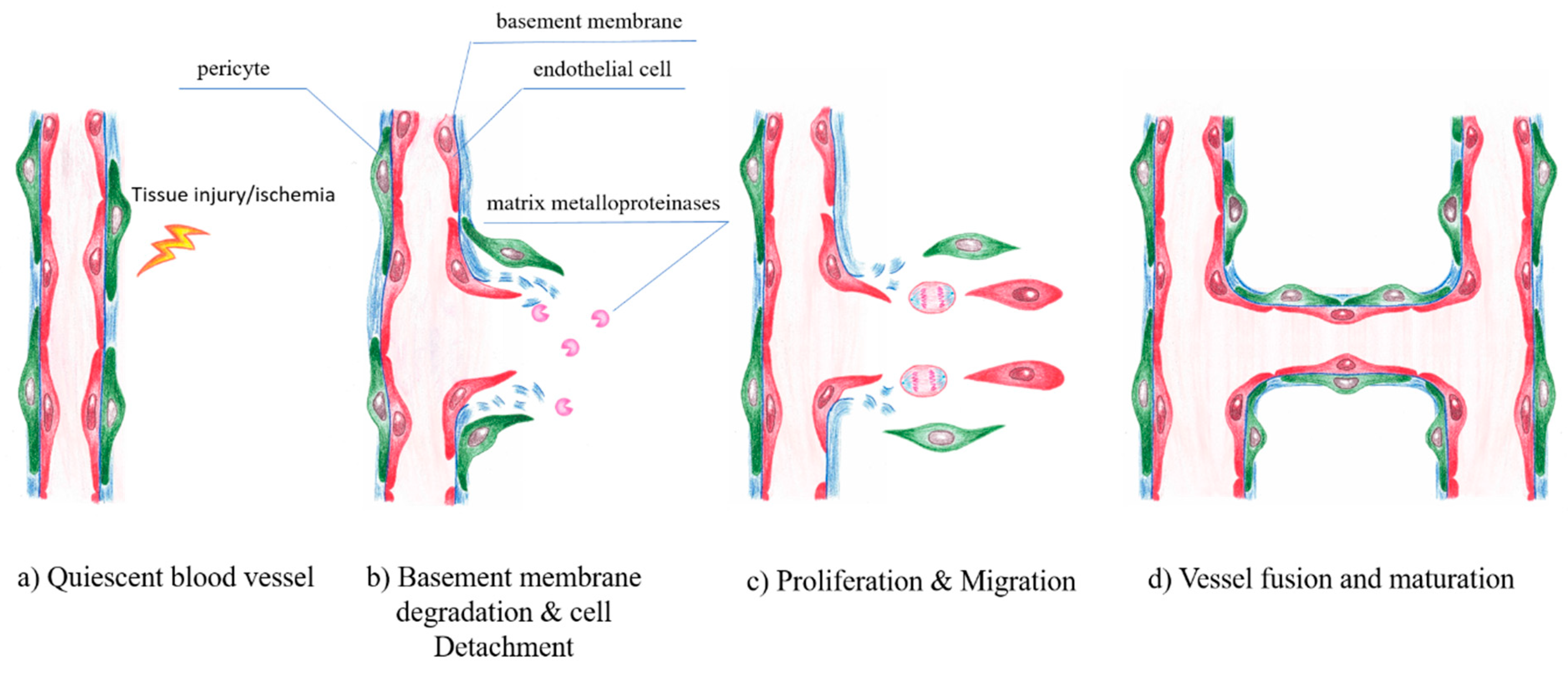
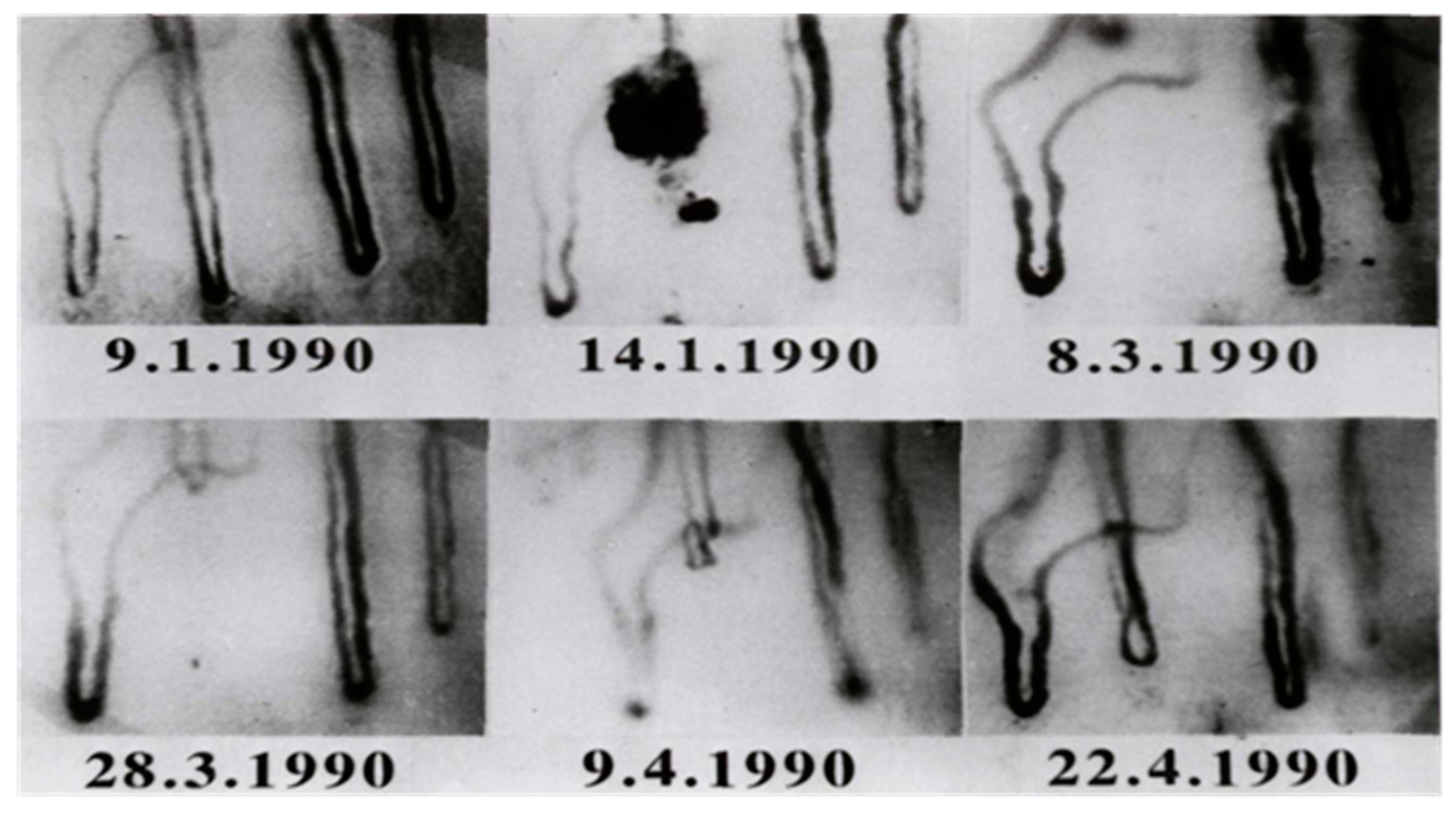
7. Endothelial Cells and Angiogenesis
8. Hematogenous Metastasis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gimbrone, M.A. Vascular endothelium: nature’s blood container. In Vascular endothelium in hemostasis and thrombosis; Gimbrone, M.A., Ed.; Churchill Livingstone: New York, NY, USA, 1986; pp. 1–13. [Google Scholar]
- Wolinsky, H. A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circ. Res. 1980, 47, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Cell Adherens Junction Assembly and Morphogenesis Induced by Sphingosine-1-Phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef]
- Geiger, B. A 130K protein from chicken gizzard: Its localization at the termini of microfilament bundles in cultured chicken cells. Cell 1979, 18, 193–205. [Google Scholar] [CrossRef]
- Michel, C.C.; Curry, F.E. Microvascular Permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef] [PubMed]
- McMaster, P.D.; Hudack, S. The vessels involved in hydrostatic transudation. J. Exp. Med. 1932, 55, 417–430. [Google Scholar] [CrossRef][Green Version]
- Crone, C. Permeability of single capillaries compared with results from whole-organ studies. Acta Physiol. Scand. Suppl. 1979, 463, 75–80. [Google Scholar] [PubMed]
- Krogh, A. The number and distribution of capillaries in muscles with calculations of the oxygen pressure head necessary for supplying the tissue. J. Physiol. 1919, 52, 409–415. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Monahan-Earley, R.; Dvorak, A.M.; Aird, W.C. Evolutionary origins of the blood vascular system and endothelium. J. Thromb. Haemost. 2013, 11, 46–66. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Krüger, A.; Mrowietz, C.; Lendlein, A.; Jung, F. Interaction of human umbilical vein endothelial cells (HUVEC) with platelets in vitro: Influence of platelet concentration and reactivity. Clin. Hemorheol. Microcirc. 2013, 55, 111–120. [Google Scholar] [PubMed]
- Tailor, A.; Cooper, D.; Granger, D.N. Platelet–Vessel Wall Interactions in the Microcirculation. Microcirculation 2005, 12, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A.; vane, J.R. Human arterial and venous tissues generate prostacyclin (prostaglandin X), a potent inhibitor of platelet aggregation. Lancet 1977, 1, 18–20. [Google Scholar] [CrossRef]
- Weksler, B.B.; Marcus, A.J.; Jaffe, E.A. Synthesis of PGI2 (prostacyclin) by cultured human and bovine endothelial cells. Proc. Nat. Acad. Sci. USA 1977, 74, 3922–3926. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Hiley, C.R.; Dora, K.A. EDHF: Spreading the influence of the endothelium. Br. J. Pharmacol. 2011, 164, 839–852. [Google Scholar] [CrossRef]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef]
- Lubrano, V.; Balzan, S. Roles of LOX-1 in microvascular dysfunction. Microvasc. Res. 2016, 105, 132–140. [Google Scholar] [CrossRef]
- Ushiyama, A.; Kataoka, H.; Iijima, T. Glycocalix and its involvement in clinical pathophysiologies. J. Intens. Care. 2016, 4, 59. [Google Scholar] [CrossRef]
- Becker, B.F.; Chappell, D.; Bruegger, D.; Annecke, T.; Jacob, M. Therapeutic strategies targeting the endothelial glycocalyx: Acute deficits, but great potential. Cardiovasc. Res. 2010, 87, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Lipowsky, H.H. Microvascular rheology and hemodynamics. Microcirculation 2005, 12, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y. Endothelial glycocalyx as a critical signaling platform integrating the extracellular hemodynamic forces and chemical signaling. J. Cell Mol. Med. 2017, 21, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef] [PubMed]
- Potter, D.R.; Jiang, J.; Damiano, E.R. The recovery time course of the endothelial-cell glycocalyx in vivo and its implications in vitro. Circ. Res. 2009, 104, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Bruegger, D.; Rehm, M.; Stoeckelhuber, M.; Welsch, U.; Conzen, P.; Becker, B.F. The endothelial glycocalyx affords compatibility of Starling’s principle and high cardiac interstitial albumin levels. Cardiovasc. Res. 2007, 73, 575–586. [Google Scholar] [CrossRef]
- Lebel, L. Clearance of hyaluronan from the circulation. Adv. Drug Delivery Rev. 1991, 2, 221–235. [Google Scholar] [CrossRef]
- Koning, N.J.; Vonk, A.B.; Vink, H.; Boer, C. Side-by-side alterations in glycocalyx thickness and perfused microvascular density during acute microcirculatory alterations in cardiac surgery. Microcirculation 2016, 23, 69–74. [Google Scholar] [CrossRef]
- Fransson, L.-Å.; Belting, M.; Cheng, F.; Jönsson, M.; Mani, K.; Sandgren, S. Novel aspects of glypican glycobiology. Cell Mol. Life Sci. CMLS. 2004, 61, 1016–1024. [Google Scholar] [CrossRef]
- Iozzo, R.V. Proteoglycans: Structure, function, and role in neoplasia. Lab. Invest. 1985, 53, 373–396. [Google Scholar] [PubMed]
- Curry, F.E.; Adamson, R.H. Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef]
- van den Berg, B.M.; Nieuwdorp, M.; Stroes, E.S.; Vink, H. Glycocalyx and endothelial (dys) function: From mice to men. Pharmacol. Rep. 2006, 58, 75–80. [Google Scholar]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflugers Arch. 2000, 440, 653–666. [Google Scholar] [CrossRef]
- Levick, J. Capillary filtration-absorption balance reconsidered in light of dynamic extravascular factors. Exp. Physiol. 1991, 76, 825–857. [Google Scholar] [CrossRef]
- Lipowsky, H.H.; Lescanic, A. Inhibition of inflammation induced shedding of the endothelial glycocalyx with low molecular weight heparin. Microvasc. Res. 2017, 112, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Tarbell, J.M.; Pahakis, M. Mechanotransduction and the glycocalyx. J. Intern. Med. 2006, 259, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Cardillo, C. Obesity, blood vessels and metabolic syndrome. Acta Physiol. (Oxf). 2011, 203, 279–286. [Google Scholar] [CrossRef]
- Reinhart, W.H. Shear-dependence of endothelial functions. Experientia 1994, 50, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Resnick, N.; Gimbrone, M.A. Hemodynamic forces are complex regulators of endothelial gene expression. FASEB J. 1995, 9, 874–882. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, Z.; Ranjan, V.; Diamond, S.L. Shear stress induction of the endothelial nitric oxide synthase gene is calcium-dependent but not calcium-activated. J. Cell Physiol. 1997, 171, 205–211. [Google Scholar] [CrossRef]
- Nerem, R.M.; Alexander, R.W.; Chappell, D.C.; Medford, R.M.; Varner, S.E.; Taylor, W.R. The study of the influence of flow on vascular endothelial biology. Am. J. Med. Sci. 1998, 316, 169–175. [Google Scholar] [PubMed]
- Chien, S. Effects of disturbed flow on endothelial cells. Ann. Biomed. Eng. 2008, 36, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.P.; Gräfe, M.; Schnittler, H.; Seiffge, D.; Mittermayer, C.; Drenckhahn, D. Induction of human vascular endothelial stress fibres by fluid shear stress. Nature 1984, 307, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Gudi, S.; Huvar, I.; White, C.R.; McKnight, N.L.; Dusserre, N.; Boss, G.R.; Frangos, J.A. Rapid activation of Ras by fluid flow is mediated by Galpha(q) and Gbetagamma subunits of heterotrimeric G proteins in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Hierck, B.P.; Van der Heiden, K.; Alkemade, F.E.; Van de Pas, S.; Van Thienen, J.V.; Groenendijk, B.C.; Bax, W.H.; Van der Laarse, A.; Deruiter, M.C.; Horrevoets, A.J.; et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn. 2008, 237, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Bergaya, S.; Murata, T.; Alp, I.F.; Bauer, M.P.; Lin, M.I.; Drab, M.; Kurzchalia, T.V.; Stan, R.V.; Sessa, W.C. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J. Clin. Invest. 2006, 116, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; del Pozo, M.A.; Chen, K.; Miao, H.; Li, Y.; Schwartz, M.A.; Shyy, J.Y.; Chien, S. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc Natl Acad Sci. USA 2001, 98, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Pahakis, M.Y.; Kosky, J.R.; Dull, R.O.; Tarbell, J.M. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 2007, 355, 228–233. [Google Scholar] [CrossRef]
- Drenckhahn, D.; Gress, T.; Franke, R.P. Vascular endothelial stress fibres: Their potential role in protecting the vessel wall from rheological damage. Klin Wochenschr. 1986, 64, 986–988. [Google Scholar] [PubMed]
- Jin, Z.G.; Ueba, H.; Tanimoto, T.; Lungu, A.O.; Frame, M.D.; Berk, B.C. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ. Res. 2003, 93, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luskinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, M.; Simionescu, N.; Santoro, F.; Palade, G.E. Differentiated microdomains of the luminal plasmalemma of murine muscle capillaries: Segmental variations in young and old animals. J. Cell Biol. 1985, 100, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, M.E.; Printz, M.P. Sites of prostaglandin synthesis in the bovine heart and isolated bovine coronary microvessels. Circ. Res. 1981, 49, 1152–1163. [Google Scholar] [CrossRef]
- Hauser, S.; Jung, F.; Pietzsch, J. Human Endothelial Cell Models in Biomaterial Research. Trends Biotechnol. 2017, 35, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R. Human pulmonary endothelial cells in culture. Activities of cells from arteries and cells from veins. J. Clin. Invest. 1980, 65, 841–850. [Google Scholar] [CrossRef]
- Michiels, C. Endothelial Cell Functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Gori, T. Endothelial Function: A Short Guide for the Interventional Cardiologist. Int. J. Mol. Sci. 2018, 19, 3838. [Google Scholar] [CrossRef]
- Miura, H.; Gutterman, D.D. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P-450 monooxygenase and Ca2+ activated K+ channels. Circ. Res. 1998, 83, 501–507. [Google Scholar] [CrossRef]
- Goto, K.; Ohtsubo, T.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. Int. J. Mol. Sci. 2018, 19, 315. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.C.; Tan, C.S.; Ch’ng, Y.S.; Yeap, Z.Q.; Ng, C.H.; Yam, M.F. Overview of the Microenvironment of Vasculature in Vascular Tone Regulation. Int. J. Mol. Sci. 2018, 19, 120. [Google Scholar]
- Fleming, I.; Bauersachs, J.; Busse, R. Paracrine functions of the coronary vascular endothelium. Mol. Cell. Biochem. 1996, 157, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li P-LRitter, J.K. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef] [PubMed]
- Komori, K.; Vanhoutte, P.M. Endothelium-derived hyperpolarizing factor. Blood Vessels 1990, 27, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Zeldin, D.C. Epoxygenase pathways of arachidonic acid metabolism. J. Biol. Chem. 2001, 276, 36059–36062. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Schulz, C.; Blaschke, F.; Muller, D.N.; Mrowietz, C.; Franke, R.P.; Lendlein, A.; Schunck, W.H. Effect of cytochrome P450-dependent epoxyeicosanoids on Ristocetin-induced thrombocyte aggregation. Clin. Hemorheol. Microcirc. 2012, 52, 403–416. [Google Scholar] [PubMed]
- Chen, J.K.; Capdevila, J.; Harris, R.C. Overexpression of C-terminal Src kinase blocks 14,15-epoxyeicosatrienoic acid-induced tyrosine phosphorylation and mitogenesis. J. Biol. Chem. 2000, 275, 13789–13792. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kalish, B.T.; Huang, S.; Bielenberg, D.R.; Le, H.D.; Yang, J.; Edin, M.L.; Lee, C.R.; Benny, O.; Mudge, D.K.; et al. Epoxyeicosanoids promote organ and tissue regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13528–13533. [Google Scholar] [CrossRef]
- Sun, J.; Sui, X.; Bradbury, J.A.; Zeldin, D.C.; Conte, M.S.; Liao, J.K. Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ. Res. 2002, 90, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Ruan, X.L.; Dai, J.; Yang, S.X.; Graham, L.; Zeldin, D.C.; Liao, J.K. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 2001, 276, 15983–15989. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lin, L.; Jiang, J.; Wang, Y.; Lu, Z.Y.; Bradbury, J.A.; Lih, F.B.; Wang, D.W.; Zeldin, D.C. Up-regulation of endothelial nitric-oxide synthase by endothelium-derived hyperpolarizing factor involves mitogen-activated protein kinase and protein kinase C signaling pathways. J. Pharmacol. Exp. Ther. 2003, 307, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Luksha, L.; Luksha, N.; Kublickas, M.; Nisell, H.; Kublickiene, K. Diverse mechanisms of endothelium-derived hyperpolarizing factor-mediated dilatation in small myometrial arteries in normal human pregnancy and preeclampsia. Biol. Reprod. 2010, 83, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, H.; Hattori, Y.; Fukao, M.; Sato, A.; Liu, M.; Sakuma, I.; Kitabatake, A.; Kanno, M. Relaxation in different-sized rat blood vessels mediated by endothelium-derived hyperpolarizing factor: Importance of processes mediating precontractions. J. Vasc. Res. 1999, 36, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Sun, D.; Smith, C.J.; Connetta, J.A.; Shesely, E.G.; Koller, A.; Kaley, G. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H762–H768. [Google Scholar] [CrossRef]
- Feletou, M.; Vanhoutte, P.M. Endothelium-derived hyperpolarizing factor: Where are we now? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1215–1225. [Google Scholar] [CrossRef]
- Félétou, M.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988, 93, 515–524. [Google Scholar] [CrossRef]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signaling. Acta Physiol. 2017, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. TE Endothelium-dependent contractions: When a good guy turns bad! J. Physiol. 2008, 15, 5295–5304. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, R.M.; Yanagisawa, M. Endothelin system: The double-edged sword in health and disease. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 851–876. [Google Scholar] [CrossRef] [PubMed]
- Rubanyi, G.M.; Botelho, L.H. Endothelins. FASEB J. 1991, 5, 2713–2720. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.M.; Nawroth, P.P. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J. Exp. Med. 1986, 163, 740–745. [Google Scholar]
- Colucci, M.; Balconi, G.; Lorenzet, R.; Pietra, A.; Locati, D.; Donati, M.B.; Semeraro, N. Cultured human endothelial cells generate tissue factor in response to endotoxin. J. Clin. Invest. 1983, 71, 1893–1896. [Google Scholar] [CrossRef]
- Lyberg, T.; Qaldal, K.S.; Evensen, S.A.; Prydz, H. Cellular cooperation in endothelial cell thromboplastin synthesis. Br. J. Haematol. 1983, 53, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.L.; Bang, N.U.; Esmon, C.T.; Esmon, N.L. The thrombogenic endothelial cells. Two endotoxin-mediated mechanisms [abstr]. Clin Res. 1985, 33, 35Oa. [Google Scholar]
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Cotran, R.S.; Gimbrone, M.A. Interleukin-1 biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J. Exp. Med. 1984, 60, 618–623. [Google Scholar] [CrossRef]
- Galdal, K.S.; Lyberg, T.; Evensen, S.A.; Nllson, E.; Prydz, H. Thrombin induces thromboplastin synthesis in cultured vascular endothelial cells. Thromb. Haemost. 1985, 54, 373–376. [Google Scholar] [CrossRef]
- Ramasamy, I. Inherited bleeding disorders: Disorders of platelet adhesion and aggregation. Crit. Rev. Oncol. Hematol. 2004, 49, 1–35. [Google Scholar] [CrossRef]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [PubMed]
- de Graaf, J.C.; Banga, J.D.; Moncada, S.; Palmer, R.M.; de Groot, P.G.; Sixma, J.J. Nitric oxide functions as an inhibitor of platelet adhesion under flow conditions. Circulation 1992, 85, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D.; Carleton, J.S.; Gordon, J.L. Metabolism of adenine nucleotides by ectoenzymes of vascular endothelial and smooth muscle cells in culture. Biochem. J. 1980, 190, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Michal, F.; Thorp, R.H. Enhanced adenosine inhibition of platelet aggregation in the presence of cardiac glycosides. Nature 1966, 209, 208–209. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.E. Thrombomodulin structure and function. Thromb. Haemost. 1997, 78, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.M.; Esposito, C.; Gerlach, H.; Gerlach, M.; Ryan, J.; Handley, D.; Nawroth, P. Endothelium and regulation of coagulation. Diabetes Care. 1991, 14, 160–166. [Google Scholar] [CrossRef]
- Esmon, C.T. The endothelial cell protein C receptor. Thromb. Haemost. 2000, 83, 639–643. [Google Scholar] [CrossRef]
- Esmon, C.T. Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J. 1995, 9, 946–955. [Google Scholar] [CrossRef]
- Kato, H. Regulation of functions of vascular wall cells by tissue factor pathway inhibitor: Basic and clinical aspects. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 539–548. [Google Scholar] [CrossRef]
- Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Invest. 2012, 122, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Crit. Care Med. 2003, 31, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Kuebler, W.M. Normal endothelium. In The Vascular Endothelium I; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–40. [Google Scholar]
- Lin, F.J.; Tsai, M.J.; Tsai, S.Y. Artery and vein formation: A tug of war between different forces. EMBO Rep. 2007, 8, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef] [PubMed]
- Carver, L.A.; Schnitzer, J.E. Proteomic mapping of endothelium and vascular targeting in vivo. In Endothelial Biomedicine; William, C., Aird, M.D., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 881–897. [Google Scholar]
- Franke, R.P.; Fuhrmann, R.; Hiebl, B.; Jung, F. Influence of various radiographic contrast media on the buckling of endothelial cells. Microvasc. Res. 2008, 76, 110–113. [Google Scholar] [CrossRef]
- Jung, F. From hemorheology to microcirculation and regenerative medicine: Fåhraeus Lecture 2009. Clin. Hemorheol. Microcirc. 2010, 45, 79–99. [Google Scholar] [PubMed]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.F. An evolving new paradigm: Endothelial cells – conditional innate immune cells. J. Hematol. Oncol. 2013, 6, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, N.; Riese, K.H.; Freudenberg, M.A. The vascular endothelial system. Structure-function-pathology-reaction to endotoxin shock-methods of investigation. Veröff. Pathol. 1983, 120, 1–114. [Google Scholar]
- Thorgeirsson, G.; Robertson, A.L., Jr. The vascular endothelium-pathobiologic significance. Am. J. Pathol. 1978, 93, 803–848. [Google Scholar]
- Gebrane-Younes, J.; Drouet, L.; Caen, J.P.; Orcel, L. Heterogeneous distribution of Weibel-Palade bodies and von Willebrand factor along the porcine vascular tree. Am. J. Pathol. 1991, 139, 1471–1484. [Google Scholar]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Mackie, K.; Lai, Y.; Nairn, A.C.; Greengard, P.; Pitt, B.R.; Lazo, J.S. Protein phosphorylation in cultured endothelial cells. J. Cell Physiol. 1986, 128, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Snopko, R.; Guffy, T.; Rafelson, M.; Hall, E. Serum stimulation of prostacyclin synthesis in aortically, venously and microvascularly derived endothelial cells. Clin. Physiol. Biochem. 1987, 5, 70–76. [Google Scholar] [PubMed]
- Franke, R.P.; Fuhrmann, R.; Mrowietz, C.; Hiebl, B.; Jung, F. Do radiographic contrast media (Iodixanol or Iomeprol) induce a perturbation of human arterial and/or venous endothelial cells in vitro on extracellular matrix? Clin. Hemorheol. Microcirc. 2012, 50, 49–54. [Google Scholar] [PubMed]
- Franke, R.P.; Fuhrmann, R.; Hiebl, B.; Jung, F. Influence of radiographic contrast media (iodixanol und iomeprol) on the morphology of human arterial and venous endothelial cells on extracellular matrix in vitro. Clin Hemorheol Microcirc. 2011, 48, 41–56. [Google Scholar] [PubMed]
- Defilippi, P.; Vanhinsbergh, V.; Bertolotto, A.; Rossino, P.; Silengo, L.; Tarone, G. Differential distribution and modulation of expression of alpha1/beta1 integrin on human endothelial-cells. J Cell Biol. 1991, 114, 855–863. [Google Scholar] [CrossRef]
- Keuschnigg, J.; Henttinen, T.; Auvinen, K.; Karikoski, M.; Salmi, M.; Jalkanen, S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood 2009, 114, 478–484. [Google Scholar] [CrossRef]
- Hoepken, S.; Fuhrmann, R.; Jung, F.; Franke, R.P. Shear resistance of human umbilical endothelial cells on different materials covered with or without extracellular matrix: Controlled in-vitro study. Clin Hemorheol Microcirc. 2009, 43, 157–166. [Google Scholar]
- Krüger-Genge, A.; Schulz, C.; Kratz, K.; Lendlein, A.; Jung, F. Comparison of two substrate materials used as negative control in endothelialization studies: Glass versus polymeric tissue culture plate. Clin Hemorheol Microcirc. 2018, 69, 437–445. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Munaron, L.; Fiorio Pla, A. Endothelial calcium machinery and angiogenesis: Understanding physiology to interfere with pathology. Curr Med Chem. 2009, 16, 4691–4703. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr Med Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Franke, R.P.; Mrowietz, C.; Wolf, S.; Kiesewetter, H. Capillary occlusion and secondary angiogenesis in a patient with Raynaud’s phenomenon. J Vasc Res. 1992, 29, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Blocki, A.; Beyer, S.; Jung, F.; Raghunath, M. The controversial origin of pericytes during angiogenesis - Implications for cell-based therapeutic angiogenesis and cell-based therapies. Clin Hemorheol Microcirc. 2018, 69, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Koch, M.; Lee, Y.H.; Jung, F.; Blocki, A. An In Vitro Model of Angiogenesis during Wound Healing Provides Insights into the Complex Role of Cells and Factors in the Inflammatory and Proliferation Phase. Int J Mol Sci. 2018, 19, 2913. [Google Scholar] [CrossRef]
- Blocki, A.; Wang, Y.; Koch, M.; Goralczyk, A.; Beyer, S.; Agarwal, N.; Lee, M.; Moonshi, S.; Dewavrin, J.Y.; Peh, P.; et al. Sourcing of an alternative pericyte-like cell type from peripheral blood in clinically relevant numbers for therapeutic angiogenic applications. Mol Ther. 2015, 23, 510–522. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Kobayashi, H.; Lin, P.C. Angiogenesis links chronic inflammation with cancer. Methods Mol Biol. 2009, 511, 185–191. [Google Scholar]
- Kundu, J.K.; Surh, Y.J. Inflammation: Gearing the journey to cancer. Mutat Res. 2008, 659, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Ma, J.X. Angiogenesis in diabetes and obesity. Rev Endocr Metab Disord. 2015, 16, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ribot, J.; Caliaperoumal, G.; Paquet, J.; Boisson-Vidal, C.; Petite, H.; Anagnostou, F. Type 2 diabetes alters mesenchymal stem cell secretome composition and angiogenic properties. J Cell Mol Med. 2017, 21, 349–363. [Google Scholar] [CrossRef]
- Hughes, C.C. Endothelial-stromal interactions in angiogenesis. Curr Opin Hematol. 2008, 15, 204–209. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef]
- Tang, H.; Lee, M.; Kim, E.H.; Bishop, D.; Rodgers, G.M. siRNA-knockdown of ADAMTS-13 modulates endothelial cell angiogenesis. Microvasc Res. 2017, 113, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Rickert, D.; Lendlein, A.; Kelch, S.; Moses, M.A.; Franke, R.P. Expression of MMPs and TIMPs in primary epithelial cell cultures of the upper aerodigestive tract seeded on the surface of a novel polymeric biomaterial. Clin Hemorheol Microcirc. 2005, 32, 117–128. [Google Scholar]
- Rhodes, J.M.; Simons, M. The extracellular matrix and blood vessel formation: Not just a scaffold. J Cell Mol Med. 2007, 11, 176–205. [Google Scholar] [CrossRef]
- Jung, F.; Franke, R.P.; Mrowietz, C.; Wolf, S.; Kiesewetter, H.; Wenzel, E. Capillary occlusion and secondary angiogenesis: Case report. Int J Microcirc Clin Exp 1992, 11, 167–170. [Google Scholar]
- Baer-Suryadinata, C.; Bollinger, A. Transcapillary diffusion of Na-fluorescein measured by a ‘large window technique’ in skin areas of the forefoot. Int J Microcirc Clin Exp. 1985, 4, 217–228. [Google Scholar]
- Eelen, G.; Cruys, B.; Welti, J.; De Bock, K.; Carmeliet, P. Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab. 2013, 24, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Ghesquiere, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Phng, L.K.; Gerhardt, H. Angiogenesis: A team effort coordinated by notch. Dev. Cell. 2009, 16, 196–208. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Zhao, K.; Dammann, P.; Keyvani, K.; Kreitschmann-Andermahr, I.; Sure, U.; Zhu, Y. EphB4 forward signaling mediates angiogenesis caused by CCM3/PDCD10-ablation. J Cell Mol Med. 2017, 21, 1848–1858. [Google Scholar] [CrossRef]
- Bentley, K.; Jones, M.; Cruys, B. Predicting the future: Towards symbiotic computational and experimental angiogenesis research. Exp Cell Res. 2013, 319, 1240–1246. [Google Scholar] [CrossRef]
- Kiriakidis, S.; Henze, A.T.; Kruszynska-Ziaja, I.; Skobridis, K.; Theodorou, V.; Paleolog, E.M.; Mazzone, M. Factor-inhibiting HIF-1 (FIH-1) is required for human vascular endothelial cell survival. FASEB J. 2015, 29, 2814–2827. [Google Scholar] [CrossRef]
- Anderson, J.M.; McNally, A.K. Biocompatibility of implants: Lymphocyte/ macrophage interactions. Semin Immunopathol. 2011, 33, 221–233. [Google Scholar] [CrossRef]
- Andrade, S.P.; Ferreira, M.A. The Sponge Implant Model of Angiogenesis. Methods Mol Biol. 2016, 1430, 333–343. [Google Scholar]
- DiEgidio, P.; Friedman, H.I.; Gourdie, R.G.; Riley, A.E.; Yost, M.J.; Goodwin, R.L. Biomedical implant capsule formation: Lessons learned and the road ahead. Ann Plast Surg. 2014, 73, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, R.; Jung, F. The pathology of the foreign body reaction against biomaterials. J. Biomed. Mater. Res. A 2017, 105, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Ullm, S.; Krüger, A.; Tondera, C.; Gebauer, T.P.; Neffe, A.T.; Lendlein, A.; Jung, F.; Pietzsch, J. Biocompatibility and inflammatory response in vitro and in vivo to gelatin-based biomaterials with tailorable elastic properties. Biomaterials 2014, 35, 9755–9766. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.M. Tumor Metastasis in the Microcirculation. Adv Exp Med Biol. 2018, 1097, 201–218. [Google Scholar] [PubMed]
- Hekimian, K.; Meisezahl, S.; Trompelt, K.; Rabenstein, C.; Pachmann, K. Epithelial cell dissemination and re-adhesion: Analysis of factors contributing to metastasis formation in breast cancer. ISRN Oncol. 2012, 2012, 601810. [Google Scholar] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. https://doi.org/10.3390/ijms20184411
Krüger-Genge A, Blocki A, Franke R-P, Jung F. Vascular Endothelial Cell Biology: An Update. International Journal of Molecular Sciences. 2019; 20(18):4411. https://doi.org/10.3390/ijms20184411
Chicago/Turabian StyleKrüger-Genge, Anne, Anna Blocki, Ralf-Peter Franke, and Friedrich Jung. 2019. "Vascular Endothelial Cell Biology: An Update" International Journal of Molecular Sciences 20, no. 18: 4411. https://doi.org/10.3390/ijms20184411
APA StyleKrüger-Genge, A., Blocki, A., Franke, R.-P., & Jung, F. (2019). Vascular Endothelial Cell Biology: An Update. International Journal of Molecular Sciences, 20(18), 4411. https://doi.org/10.3390/ijms20184411