Gene Therapy for ALS—A Perspective

Abstract
1. Introduction
2. ALS—A Lethal Disease with No Cure
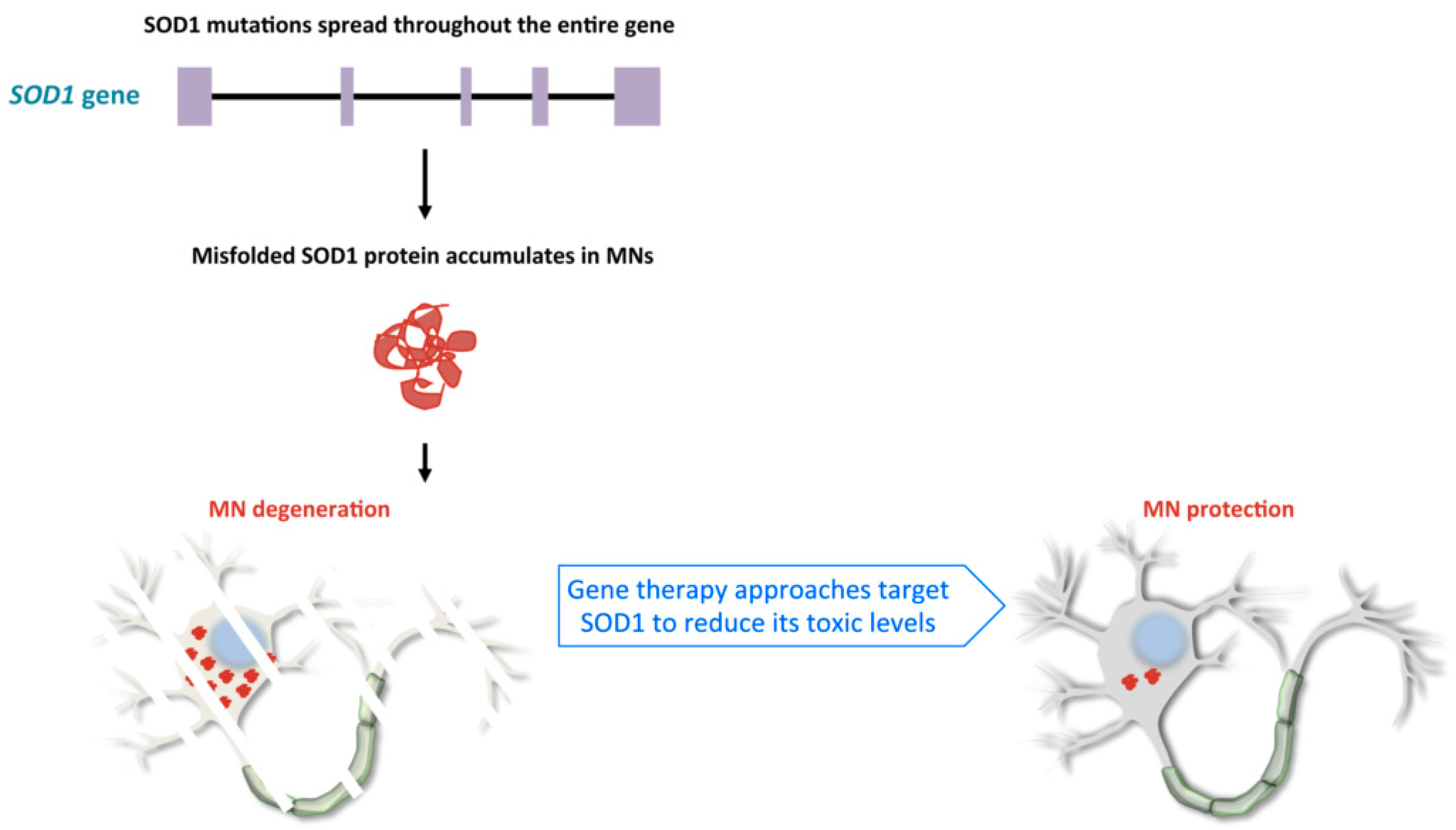
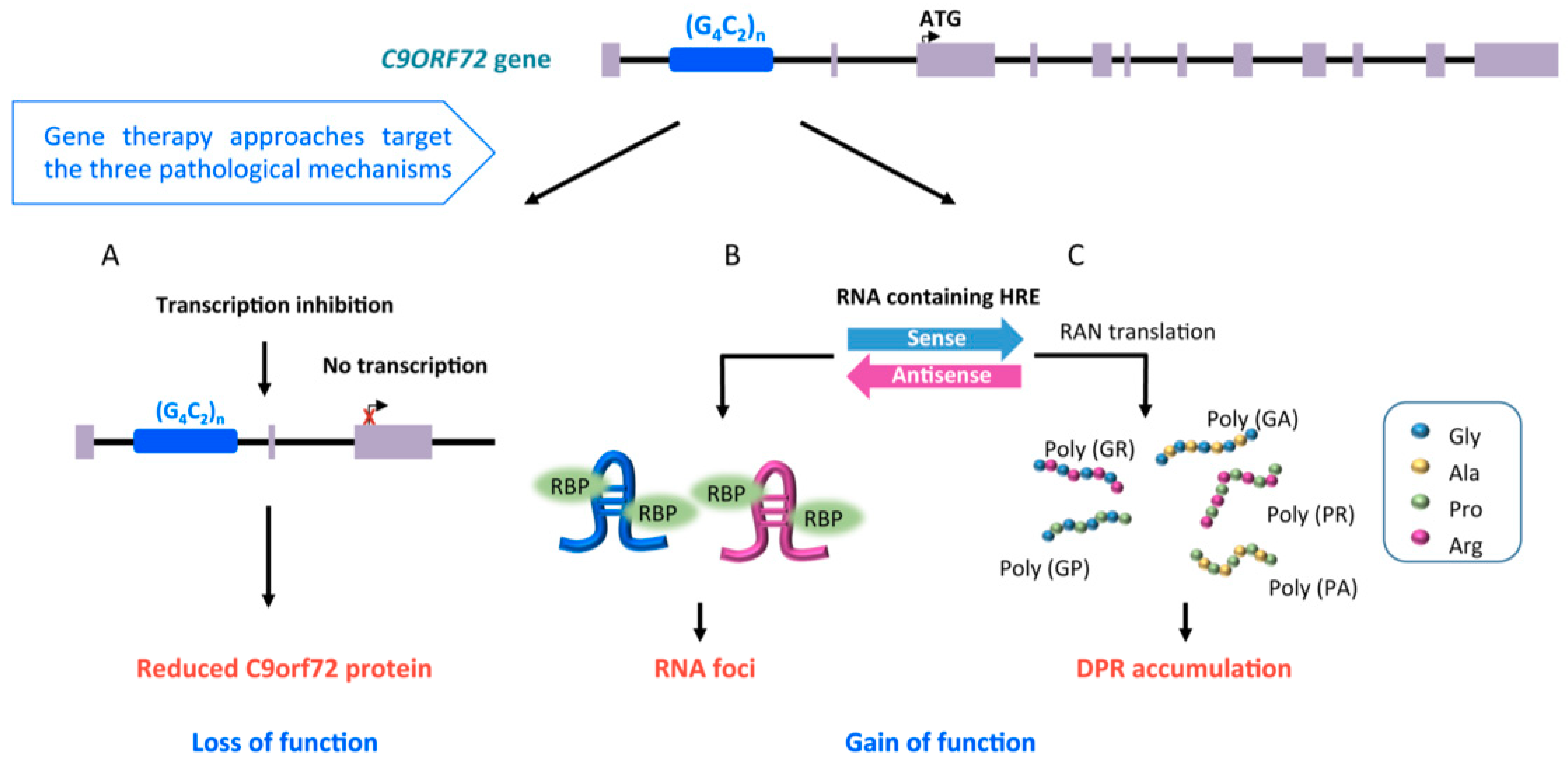
2.1. Familial Cases of ALS, Pathological Mechanisms of SOD1 and C9orf72 Mutations
2.2. Mouse Models for SOD1 and C9orf72-ALS
3. Non-Vector-Based Gene Therapy
3.1. Antisense Oligonucleotides in fALS
3.2. RNA Interference in fALS
4. Viral Vectors for MND Gene Therapy
4.1. Gene Therapy Mediated by LV
4.2. Gene Therapy Mediated by AAV
5. Genome Editing for fALS
6. Delivery of Neurotrophic Factors as Potential Treatment for All ALS Cases
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAP | Assembly-Activating Protein |
| AAV | Adeno-Associated virus |
| ALS | Amyotrophic Lateral Sclerosis |
| ASO | Antisense oligonucleotides |
| B6.SOD1-G93A | SOD1-G93A mouse model backcrossed the to the C57BL/6J |
| BAC | Bacterial artificial chromosome |
| BDNF | Brain derived neurotrophic factor |
| C9-100-1000 | BAC C9orf72 mice harboring from 100 to 1000 repeats |
| C9-450 | BAC C9orf72 mice harboring 450 repeats |
| C9-500 | BAC C9orf72 mice harboring 500 repeats |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CHMP | Committee for Medicinal Products for Human Use |
| CNS | Central nervous system |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CSF | Cerebrospinal fluid |
| DPRs | Dipeptide repeat proteins |
| EIAV | Equine infectious anaemia virus |
| EMA | European Medicines Agency |
| fALS | Familial ALS |
| FDA | Food and Drug Administration |
| FTD | Frontotemporal dementia |
| FUS | Fused in sarcoma |
| G4C2 | GGGGCC |
| G93A | single amino acid substitution of glycine to alanine at codon 93 of the human SOD1 |
| GA | Glycine–alanine |
| GDNF | Glial cell line derived neurotrophic factor |
| GP | Glycine–proline |
| GR | Glycine–arginine |
| GWAS | Genome-wide association studies |
| HiRet | Highly-efficient retrograde gene transfer |
| HIV | Human immunodeficiency virus |
| HRE | Hexanucleotide repeat expansion |
| IGF-1 | Insulin-like growth factor 1 |
| iPSC | Induced pluripotent stem cells |
| ITRs | Inverted terminal repeats |
| IV | Intravenous |
| LTR | Long terminal repeats |
| LV | Lentiviral vectors |
| miR | microRNA |
| MN | Motor neurons |
| MND | Motor neuron disease |
| MNDs | Motor neuron disorders |
| NeuRet | Neuron-specific retrograde gene transfer |
| NMD | Nonsense Mediated Decay |
| PA | Proline–alanine |
| PR | Proline–arginine |
| RAN | Repeat-associated non-AUG-dependent translation |
| RBPs | RNA-binding proteins |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| RNase H | Ribonuclease H |
| RVG | Rabies virus envelope glycoprotein |
| sALS | Sporadic ALS |
| Sc | Self-complementary |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| SMA | Spinal Muscular Atrophy |
| SMN | Survival of motor neuron |
| SOD1 | Cu/Zn superoxide dismutase 1 enzyme |
| SOD1-G93A | SOD1 mouse model in the mixed B6SJL genetic backgroundexpressing multiple copies of the human SOD1 transgene, carrying a single amino acid substitution of glycine to alanine at codon 93 |
| Ss | Single-stranded |
| TDP-43 | TAR DNA binding protein of 43 kDa |
| VEGF | Vascular endothelial growth factor |
| VSV-G | Vesicular stomatitis virus glycoprotein |
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Statland, J.M.; Barohn, R.J.; McVey, A.L.; Katz, J.S.; Dimachkie, M.M. Patterns of Weakness, Classification of Motor Neuron Disease, and Clinical Diagnosis of Sporadic Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 735–748. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Hogden, A.; Foley, G.; Henderson, R.D.; James, N.; Aoun, S.M. Amyotrophic lateral sclerosis: Improving care with a multidisciplinary approach. J. Multidiscip. Healthc. 2017, 10, 205–215. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Tosolini, A.P.; Sleigh, J.N. Motor Neuron Gene Therapy: Lessons from Spinal Muscular Atrophy for Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Smith, R.A. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clinic. Investig. 2006, 116, 2290–2296. [Google Scholar] [CrossRef]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part II. Gene therapy strategies and applications. Discov. Med. 2014, 18, 151–161. [Google Scholar]
- Goyenvalle, A. Gene and splicing therapies for neuromuscular diseases. Front. Biosci. 2015, 20, 1190–1233. [Google Scholar] [CrossRef]
- Choong, C.-J.; Baba, K.; Mochizuki, H. Gene therapy for neurological disorders. Expert Opin. Biol. Ther. 2016, 16, 143–159. [Google Scholar] [CrossRef]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: part I. Gene delivery technologies. Discov. Med. 2014, 18, 67–77. [Google Scholar]
- Durymanov, M.; Reineke, J. Non-viral Delivery of Nucleic Acids: Insight into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Schoch, K.M.; Miller, T.M. Antisense oligonucleotides: Translation from mouse models to human neurodegenerative diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Method. Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Marangi, G.; Traynor, B.J. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015, 1607, 75–93. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. Incidence of Amyotrophic Lateral Sclerosis in Europe. J. Neurol. Neurosurg. Psychiatr. 2010, 81, 385–390. [Google Scholar] [CrossRef]
- Deenen, J.C.W.; Horlings, C.G.C.; Verschuuren, J.J.; Verbeek, A.L.M.; Engelen, B.G.M. van The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernán, M.A. Incidence and lifetime risk of motor neuron disease in the United Kingdom: A population-based study. Eur. J. Neurol. 2009, 16, 745–751. [Google Scholar] [CrossRef]
- Logroscino, G.; Piccininni, M. Amyotrophic Lateral Sclerosis Descriptive Epidemiology: The Origin of Geographic Difference. NED 2019, 52, 93–103. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph. Lateral. Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral. Scler. Frontotemporal Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Strong, M.J. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2010, 288, 1–12. [Google Scholar] [CrossRef]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72 -mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544. [Google Scholar] [CrossRef]
- Xiao, S.; MacNair, L.; McGoldrick, P.; McKeever, P.M.; McLean, J.R.; Zhang, M.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 78, 568–583. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Aoki, Y.; Manzano, R.; Lee, Y.; Dafinca, R.; Aoki, M.; Douglas, A.G.L.; Varela, M.A.; Sathyaprakash, C.; Scaber, J.; Barbagallo, P.; et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2017, 140, 887–897. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural. Regen. Res. 2018, 13, 2050–2054. [Google Scholar]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Heiman-Patterson, T.D.; Deitch, J.S.; Blankenhorn, E.P.; Erwin, K.L.; Perreault, M.J.; Alexander, B.K.; Byers, N.; Toman, I.; Alexander, G.M. Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. J. Neurol. Sci. 2005, 236, 1–7. [Google Scholar] [CrossRef]
- Nagai, M.; Aoki, M.; Miyoshi, I.; Kato, M.; Pasinelli, P.; Kasai, N.; Brown, R.H.; Itoyama, Y. Rats Expressing Human Cytosolic Copper–Zinc Superoxide Dismutase Transgenes with Amyotrophic Lateral Sclerosis: Associated Mutations Develop Motor Neuron Disease. J. Neurosci. 2001, 21, 9246–9254. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef]
- Shao, Q.; Liang, C.; Chang, Q.; Zhang, W.; Yang, M.; Chen, J.-F. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun. 2019, 7, 32. [Google Scholar] [CrossRef]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar] [CrossRef]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H.M. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Haché, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children with Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child. Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clinic. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.-W.; Pham, J.T.; Heusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.M.G.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9ORF72 Repeat Expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Hannon, G.J.; Rossi, J.J. Unlocking the potential of the human genome with RNA interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef]
- Manjunath, N.; Haoquan, W.; Sandesh, S.; Premlata, S. Lentiviral delivery of short hairpin RNAs. Adv. Drug. Deliv. Rev. 2009, 61, 732–745. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ding, H.; Schwarz, D.S.; Keene, A.; Affar, E.B.; Fenton, L.; Xia, X.; Shi, Y.; Zamore, P.D.; Xu, Z. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2003, 2, 209–217. [Google Scholar] [CrossRef]
- Rizvanov, A.A.; Mukhamedyarov, M.A.; Palotás, A.; Islamov, R.R. Retrogradely transported siRNA silences human mutant SOD1 in spinal cord motor neurons. Exp. Brain Res. 2009, 195, 1–4. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Watts, J.K.; Damha, M.J. Chemical Modification of siRNA. In Current Protocols in Nucleic Acid Chemistry; Beaucage, S.L., Bergstrom, D.E., Herdewijn, P., Matsuda, A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 16.3.1–16.3.22. ISBN 978-0-471-14270-6. [Google Scholar]
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.H.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227. [Google Scholar] [CrossRef]
- Scoles, D.R.; Pulst, S.M. Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol. 2018, 15, 707–714. [Google Scholar] [CrossRef]
- Prakash, S.; Malhotra, M.; Rengaswamy, V. Nonviral siRNA delivery for gene silencing in neurodegenerative diseases. Method. Mol. Biol. 2010, 623, 211–229. [Google Scholar]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 27, 10962–10967. [Google Scholar] [CrossRef]
- Azzouz, M. Gene Therapy for ALS: Progress and prospects. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1122–1127. [Google Scholar] [CrossRef]
- Blessing, D.; Déglon, N. Adeno-associated virus and lentivirus vectors: A refined toolkit for the central nervous system. Curr. Opin. Virol. 2016, 21, 61–66. [Google Scholar] [CrossRef]
- Davidson, B.L.; Breakefield, X.O. Viral vectors for gene delivery to the nervous system. Nat. Rev. Neurosci. 2003, 4, 353–364. [Google Scholar] [CrossRef]
- Escors, D.; Breckpot, K. Lentiviral Vectors in Gene Therapy: Their Current Status and Future Potential. Arch. Immunol. Ther. Exp. 2010, 58, 107–119. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Jakobsson, J.; Lundberg, C. Lentiviral Vectors for Use in the Central Nervous System. Mol. Ther. 2006, 13, 484–493. [Google Scholar] [CrossRef]
- Naldini, L.; Blömer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef]
- Hislop, J.N.; Islam, T.A.; Eleftheriadou, I.; Carpentier, D.C.J.; Trabalza, A.; Parkinson, M.; Schiavo, G.; Mazarakis, N.D. Rabies Virus Envelope Glycoprotein Targets Lentiviral Vectors to the Axonal Retrograde Pathway in Motor Neurons. J. Biol. Chem. 2014, 289, 16148–16163. [Google Scholar] [CrossRef]
- Trabalza, A.; Eleftheriadou, I.; Sgourou, A.; Liao, T.-Y.; Patsali, P.; Lee, H.; Mazarakis, N.D. Enhanced Central Nervous System Transduction with Lentiviral Vectors Pseudotyped with RVG/HIV-1gp41 Chimeric Envelope Glycoproteins. J. Virol. 2014, 88, 2877–2890. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kato, S.; Kobayashi, K. Genetic manipulation of specific neural circuits by use of a viral vector system. J. Neural. Transm. 2018, 125, 67–75. [Google Scholar] [CrossRef]
- Hirano, M.; Kato, S.; Kobayashi, K.; Okada, T.; Yaginuma, H.; Kobayashi, K. Highly Efficient Retrograde Gene Transfer into Motor Neurons by a Lentiviral Vector Pseudotyped with Fusion Glycoprotein. PLoS ONE 2013, 8, e75896. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Raoul, C.; Abbas-Terki, T.; Bensadoun, J.-C.; Guillot, S.; Haase, G.; Szulc, J.; Henderson, C.E.; Aebischer, P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005, 11, 423–428. [Google Scholar] [CrossRef]
- Ralph, G.S.; Radcliffe, P.A.; Day, D.M.; Carthy, J.M.; Leroux, M.A.; Lee, D.C.P.; Wong, L.-F.; Bilsland, L.G.; Greensmith, L.; Kingsman, S.M.; et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005, 11, 429–433. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Hudry, E.; Maguire, C.A.; Sena-Esteves, M.; Breakefield, X.O.; Grandi, P. Viral vectors for therapy of neurologic diseases. Neuropharmacology 2017, 120, 63–80. [Google Scholar] [CrossRef]
- Suzuki, M.; Svendsen, C.N. Ex Vivo Gene Therapy Using Human Mesenchymal Stem Cells to Deliver Growth Factors in the Skeletal Muscle of a Familial ALS Rat Model. Method. Mol. Biol. 2016, 1382, 325–336. [Google Scholar]
- Samulski, R.J.; Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV in clinical trials. Curr. Gene Ther. 2007, 7, 316–324. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Muruve, D.A. Immune responses to adeno-associated virus vectors. Curr. Gene Ther. 2005, 5, 323–331. [Google Scholar] [CrossRef]
- Asokan, A.; Schaffer, D.V.; Samulski, R.J. The AAV vector toolkit: Poised at the clinical crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef]
- Hester, M.; Foust, K.; Kaspar, R.; Kaspar, B. AAV as a Gene Transfer Vector for the Treatment of Neurological Disorders: Novel Treatment Thoughts for ALS. Curr. Gene Ther. 2009, 9, 428–433. [Google Scholar] [CrossRef]
- Deverman, B.E.; Ravina, B.M.; Bankiewicz, K.S.; Paul, S.M.; Sah, D.W.Y. Gene therapy for neurological disorders: Progress and prospects. Nat. Rev. Drug Discov. 2018, 17, 641–659. [Google Scholar] [CrossRef]
- Duque, S.; Joussemet, B.; Riviere, C.; Marais, T.; Dubreil, L.; Douar, A.-M.; Fyfe, J.; Moullier, P.; Colle, M.-A.; Barkats, M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009, 17, 1187–1196. [Google Scholar] [CrossRef]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef]
- Tanguy, Y.; Biferi, M.G.; Besse, A.; Astord, S.; Cohen-Tannoudji, M.; Marais, T.; Barkats, M. Systemic AAVrh10 provides higher transgene expression than AAV9 in the brain and the spinal cord of neonatal mice. Front. Mol. Neurosci 2015, 8, 36. [Google Scholar] [CrossRef]
- Gao, G.; Alvira, M.R.; Somanathan, S.; Lu, Y.; Vandenberghe, L.H.; Rux, J.J.; Calcedo, R.; Sanmiguel, J.; Abbas, Z.; Wilson, J.M. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. USA 2003, 100, 6081–6086. [Google Scholar] [CrossRef]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef]
- Shevtsova, Z.; Malik, J.M.I.; Michel, U.; Bähr, M.; Kügler, S. Promoters and serotypes: Targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp. Physiol. 2005, 90, 53–59. [Google Scholar] [CrossRef]
- von Jonquieres, G.; Mersmann, N.; Klugmann, C.B.; Harasta, A.E.; Lutz, B.; Teahan, O.; Housley, G.D.; Fröhlich, D.; Krämer-Albers, E.-M.; Klugmann, M. Glial Promoter Selectivity following AAV-Delivery to the Immature Brain. PLoS ONE 2013, 8, e65646. [Google Scholar] [CrossRef]
- Castle, M.J.; Perlson, E.; Holzbaur, E.L.; Wolfe, J.H. Long-distance Axonal Transport of AAV9 Is Driven by Dynein and Kinesin-2 and Is Trafficked in a Highly Motile Rab7-positive Compartment. Mol. Ther. 2014, 22, 554–566. [Google Scholar] [CrossRef]
- McCarty, D.M. Self-complementary AAV Vectors; Advances and Applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal-neurons and adult-astrocytes in CNS. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef]
- Valori, C.F.; Ning, K.; Wyles, M.; Mead, R.J.; Grierson, A.J.; Shaw, P.J.; Azzouz, M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010, 2, 35ra42. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-mediated Suppression of Mutant SOD1 Slows Disease Progression and Extends Survival in Models of Inherited ALS. Mol. Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef]
- Iannitti, T.; Scarrott, J.M.; Likhite, S.; Coldicott, I.R.P.; Lewis, K.E.; Heath, P.R.; Higginbottom, A.; Myszczynska, M.A.; Milo, M.; Hautbergue, G.M.; et al. Translating SOD1 Gene Silencing toward the Clinic: A Highly Efficacious, Off-Target-free, and Biomarker-Supported Strategy for fALS. Mol. Ther. Nucleic Acids 2018, 12, 75–88. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Qiu, L.; Yang, C.; Kramer, J.; Su, Q.; Guo, Y.; Brown, R.H.; Gao, G.; Xu, Z. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 668–681. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabreja, G.C.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1G93A Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H.; Sena-Esteves, M. Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H.; Mueller, C. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med. 2018, 10, eaau6414. [Google Scholar] [CrossRef]
- Ward, A.J.; Norrbom, M.; Chun, S.; Bennett, C.F.; Rigo, F. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014, 42, 5871–5879. [Google Scholar] [CrossRef]
- Biferi, M.G.; Cohen-Tannoudji, M.; Cappelletto, A.; Giroux, B.; Roda, M.; Astord, S.; Marais, T.; Bos, C.; Voit, T.; Ferry, A.; et al. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Mol. Ther. 2017, 25, 2038–2052. [Google Scholar] [CrossRef]
- Schümperli, D.; Pillai, R.S. The special Sm core structure of the U7 snRNP: Far-reaching significance of a small nuclear ribonucleoprotein. Cell. Mol. Life Sci. 2004, 61, 2560–2570. [Google Scholar] [CrossRef]
- Martier, R.; Liefhebber, J.M.; García-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Ther. Nucleic Acids 2019, 16, 26–37. [Google Scholar] [CrossRef]
- Martier, R.; Liefhebber, J.M.; Miniarikova, J.; van der Zon, T.; Snapper, J.; Kolder, I.; Petry, H.; van Deventer, S.J.; Evers, M.M.; Konstantinova, P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-nuclear Transcripts in ALS and FTD Patients. Mol. Ther. Nucleic Acids 2019, 14, 593–608. [Google Scholar] [CrossRef]
- Cai, L.; Fisher, A.L.; Huang, H.; Xie, Z. CRISPR-mediated genome editing and human diseases. Genes Dis. 2016, 3, 244–251. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef]
- Pribadi, M.; Yang, Z.; Kim, T.S.; Swartz, E.W.; Huang, A.Y.; Chen, J.A.; Dokuru, D.; Baek, J.; Gao, F.; Fua, A.T.; et al. CRISPR-Cas9 targeted deletion of the C9orf72 repeat expansion mutation corrects cellular phenotypes in patient-derived iPS cells. bioRxiv 2016, 051193. [Google Scholar] [CrossRef]
- McEachin, Z.T.; Donsante, A.; Boulis, N. Gene Therapy for the Treatment of Neurological Disorders: Amyotrophic Lateral Sclerosis. In Gene Therapy for Neurological Disorders; Manfredsson, F.P., Ed.; Springer New York: New York, NY, USA, 2016; Volume 1382, pp. 399–408. ISBN 978-1-4939-3270-2. [Google Scholar]
- Wang, L.-J.; Lu, Y.-Y.; Muramatsu, S.; Ikeguchi, K.; Fujimoto, K.; Okada, T.; Mizukami, H.; Matsushita, T.; Hanazono, Y.; Kume, A.; et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J. Neurosci. 2002, 22, 6920–6928. [Google Scholar] [CrossRef]
- Kaspar, B.K.; Lladó, J.; Sherkat, N.; Rothstein, J.D.; Gage, F.H. Retrograde Viral Delivery of IGF-1 Prolongs Survival in a Mouse ALS Model. Science 2003, 301, 839–842. [Google Scholar] [CrossRef]
- Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; Mitrophanous, K.A.; Kingsman, S.M.; Carmeliet, P.; Mazarakis, N.D. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413. [Google Scholar] [CrossRef]
- Lepore, A.C.; Haenggeli, C.; Gasmi, M.; Bishop, K.M.; Bartus, R.T.; Maragakis, N.J.; Rothstein, J.D. Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. 2007, 1185, 256–265. [Google Scholar] [CrossRef]
- Franz, C.K.; Federici, T.; Yang, J.; Backus, C.; Oh, S.S.; Teng, Q.; Carlton, E.; Bishop, K.M.; Gasmi, M.; Bartus, R.T.; et al. Intraspinal cord delivery of IGF-I mediated by adeno-associated virus 2 is neuroprotective in a rat model of familial ALS. Neurobiol. Dis. 2009, 33, 473–481. [Google Scholar] [CrossRef]
- Dodge, J.C.; Haidet, A.M.; Yang, W.; Passini, M.A.; Hester, M.; Clarke, J.; Roskelley, E.M.; Treleaven, C.M.; Rizo, L.; Martin, H.; et al. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 2008, 16, 1056–1064. [Google Scholar] [CrossRef]
- Dodge, J.C.; Treleaven, C.M.; Fidler, J.A.; Hester, M.; Haidet, A.; Handy, C.; Rao, M.; Eagle, A.; Matthews, J.C.; Taksir, T.V.; et al. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol. Ther. 2010, 18, 2075–2084. [Google Scholar] [CrossRef]
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.H.; Talbot, K.; Consortium, T.U.S.R. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef]
- Benkler, C.; Barhum, Y.; Ben-Zur, T.; Offen, D. Multifactorial Gene Therapy Enhancing the Glutamate Uptake System and Reducing Oxidative Stress Delays Symptom Onset and Prolongs Survival in the SOD1-G93A ALS Mouse Model. J. Mol. Neurosci. 2016, 58, 46–58. [Google Scholar] [CrossRef]
- Frakes, A.E.; Braun, L.; Ferraiuolo, L.; Guttridge, D.C.; Kaspar, B.K. Additive amelioration of ALS by co-targeting independent pathogenic mechanisms. Ann. Clinic. Transl. Neurol. 2017, 4, 76–86. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tools for In Vivo Gene Therapy and Their Characteristics | |||
|---|---|---|---|
| Non-viral strategies |  Antisense Oligonucleotides (ASO) | ✓ 13–25 nucleotide long, single-stranded nucleic acid (RNA or DNA) ✓ Induction of mRNA degradation through activation of RNaseH ✓ In clinical trial for SOD1-ALS and C9-ALS ✓ ASOs can be produced by plasmids encoding for small nuclear RNA particles (such as modified U7) | ASOs and siRNAs are rapidly degraded by endonucleases and require repeated invasive injection into the central nervous system for ALS treatment. When encoded by plasmids they can be delivered using viral vectors for stable transduction. |
 Small Interfering RNAs (siRNA) | ✓ 19–23 nucleotide long, double-stranded RNA ✓ Induction of mRNA degradation through nucleases activity ✓ They can be continuously produced by plasmids encoding short-hairpin RNA or artificial microRNA | ||
| Viral vector-mediated strategies |  Lentiviral (LV) vectors | ✓ Large cloning capacity (8–10 Kb) ✓ Transduction of dividing and non-dividing cells ✓ Long-term transgene expression in dividing cells | LV have a broad tropism and transduce areas close to the injection site. They integrate into the host genome and have a mutagenic risk. |
 Adeno-Associated virus (AAV) vectors | ✓ Transduction of dividing and non-dividing cells ✓ Non pathogenic ✓ Specific tropism for different cell types, according to serotypes ✓ Persistence in the cells as extra-chromosomal episomes ✓ Low risk of insertional mutagenesis | AAV have many advantages for clinical application, but they have a small cloning capacity (single stranded: about 4.7 Kb; self complementary: about 2.4 Kb). | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS—A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. https://doi.org/10.3390/ijms20184388
Cappella M, Ciotti C, Cohen-Tannoudji M, Biferi MG. Gene Therapy for ALS—A Perspective. International Journal of Molecular Sciences. 2019; 20(18):4388. https://doi.org/10.3390/ijms20184388
Chicago/Turabian StyleCappella, Marisa, Chiara Ciotti, Mathilde Cohen-Tannoudji, and Maria Grazia Biferi. 2019. "Gene Therapy for ALS—A Perspective" International Journal of Molecular Sciences 20, no. 18: 4388. https://doi.org/10.3390/ijms20184388
APA StyleCappella, M., Ciotti, C., Cohen-Tannoudji, M., & Biferi, M. G. (2019). Gene Therapy for ALS—A Perspective. International Journal of Molecular Sciences, 20(18), 4388. https://doi.org/10.3390/ijms20184388