Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression



 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
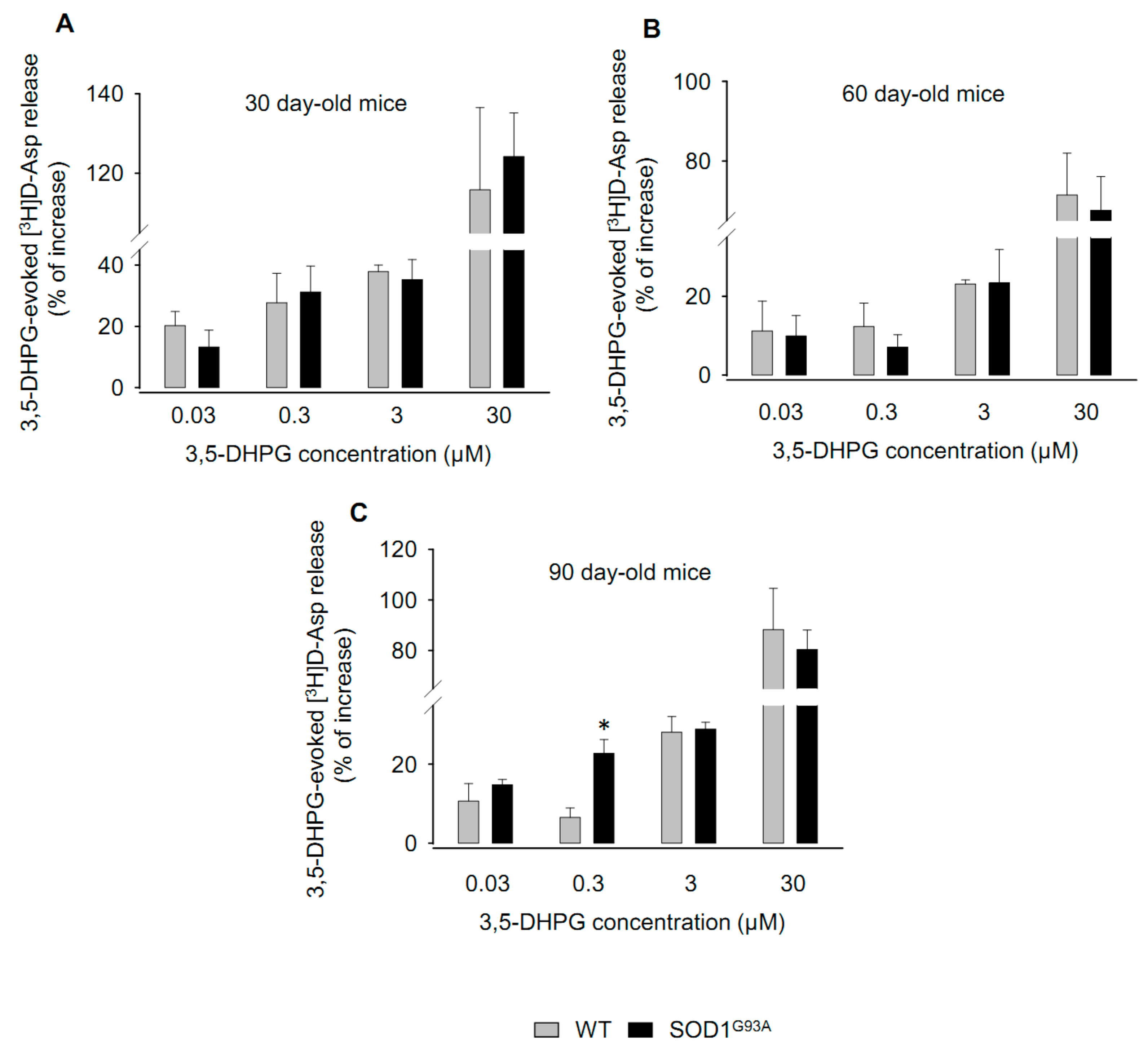
2.1. Effects of 3,5-DHPG on [3H]d-Aspartate Release during Disease Progression
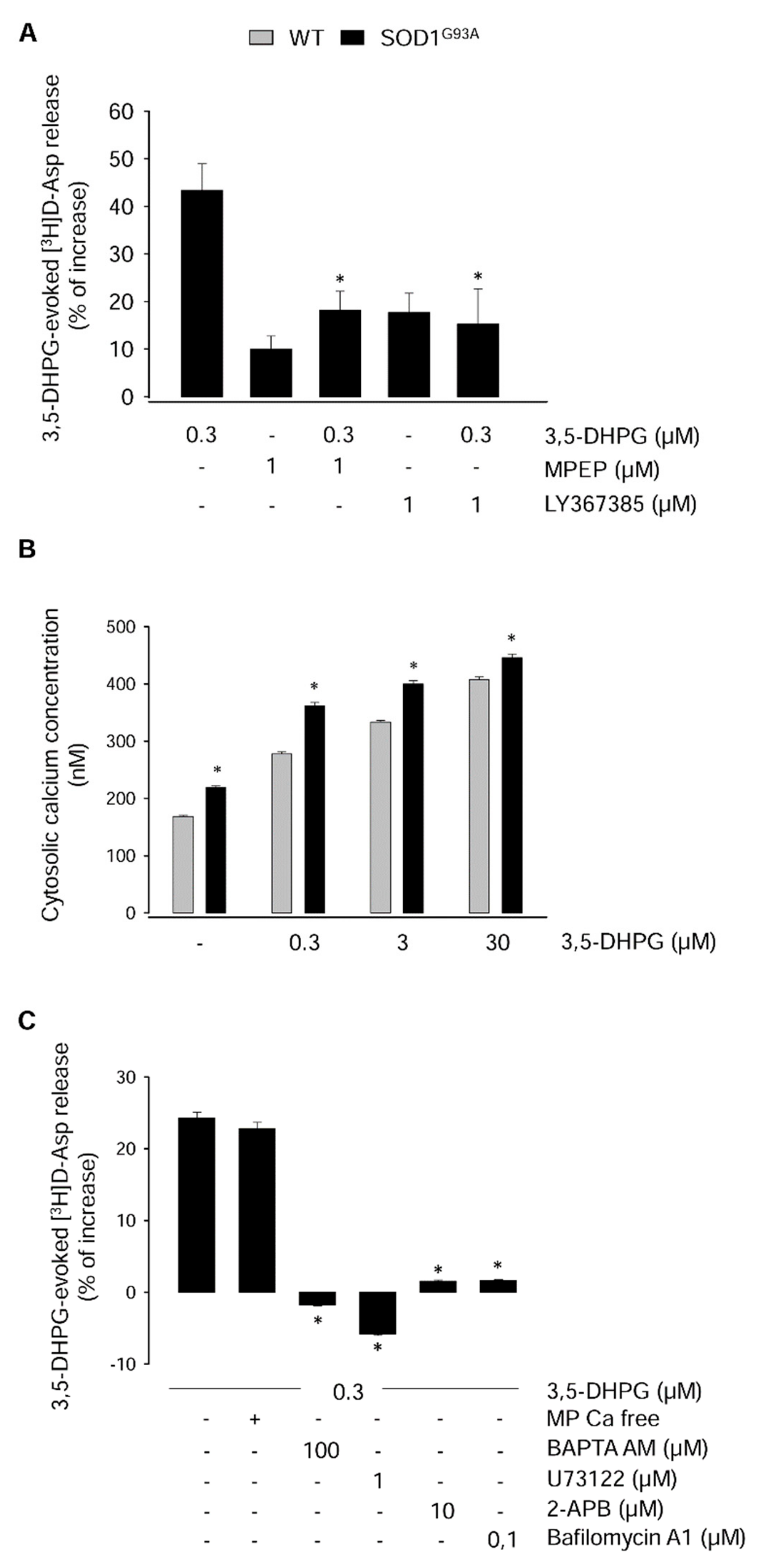
2.2. Mechanisms Underlying the Modulation of [3H]d-Aspartate Release by 3,5-DHPG in 90-Day-Old Mice
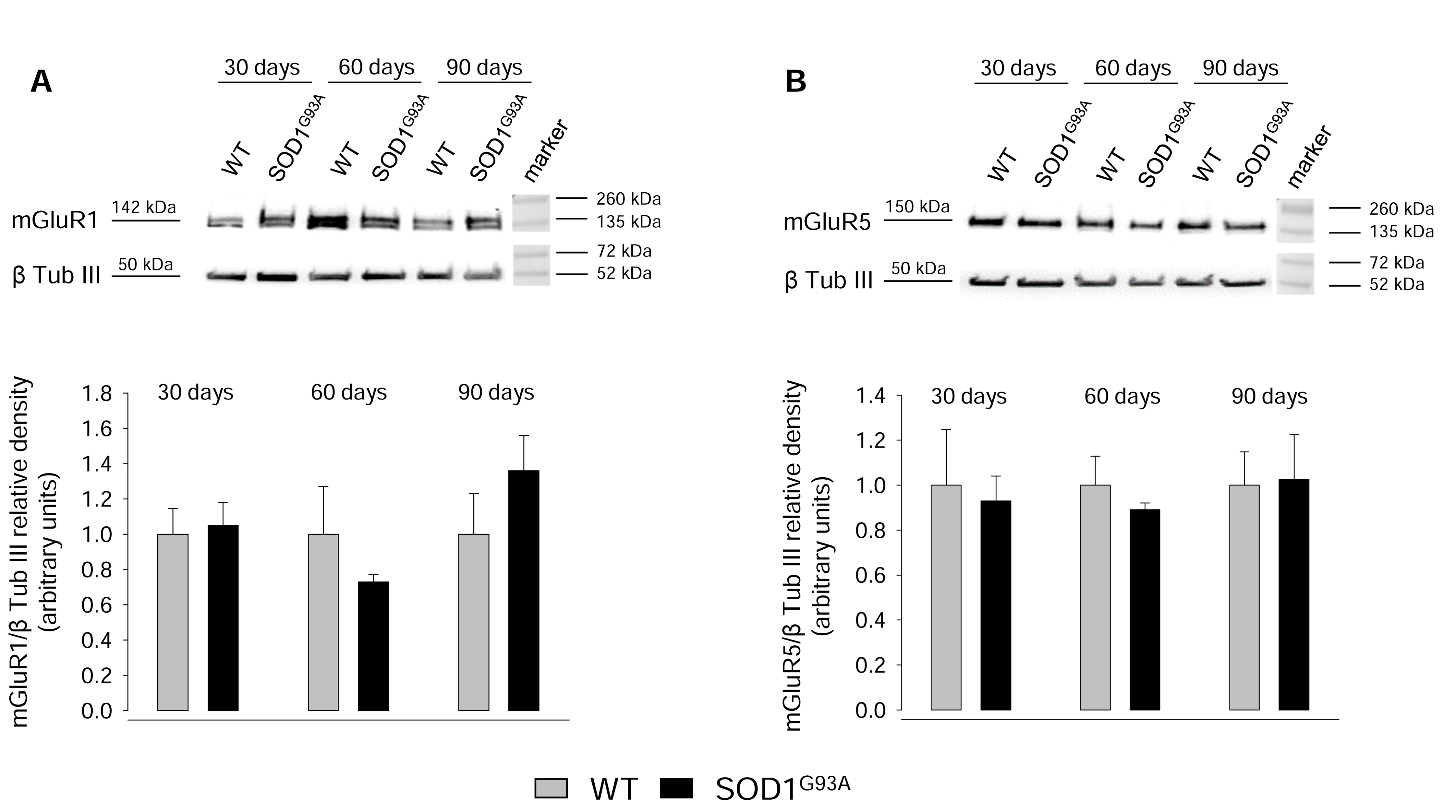
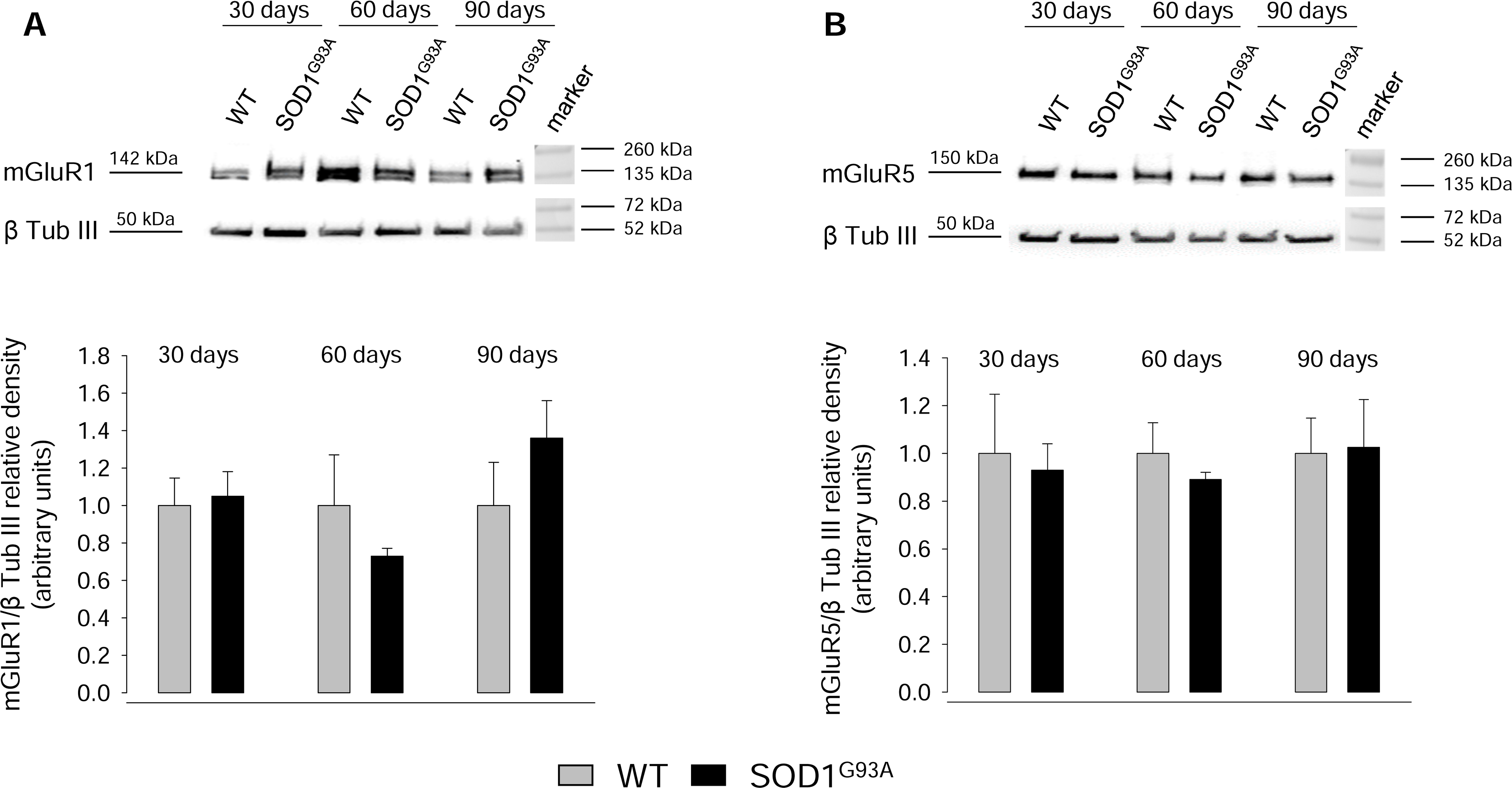
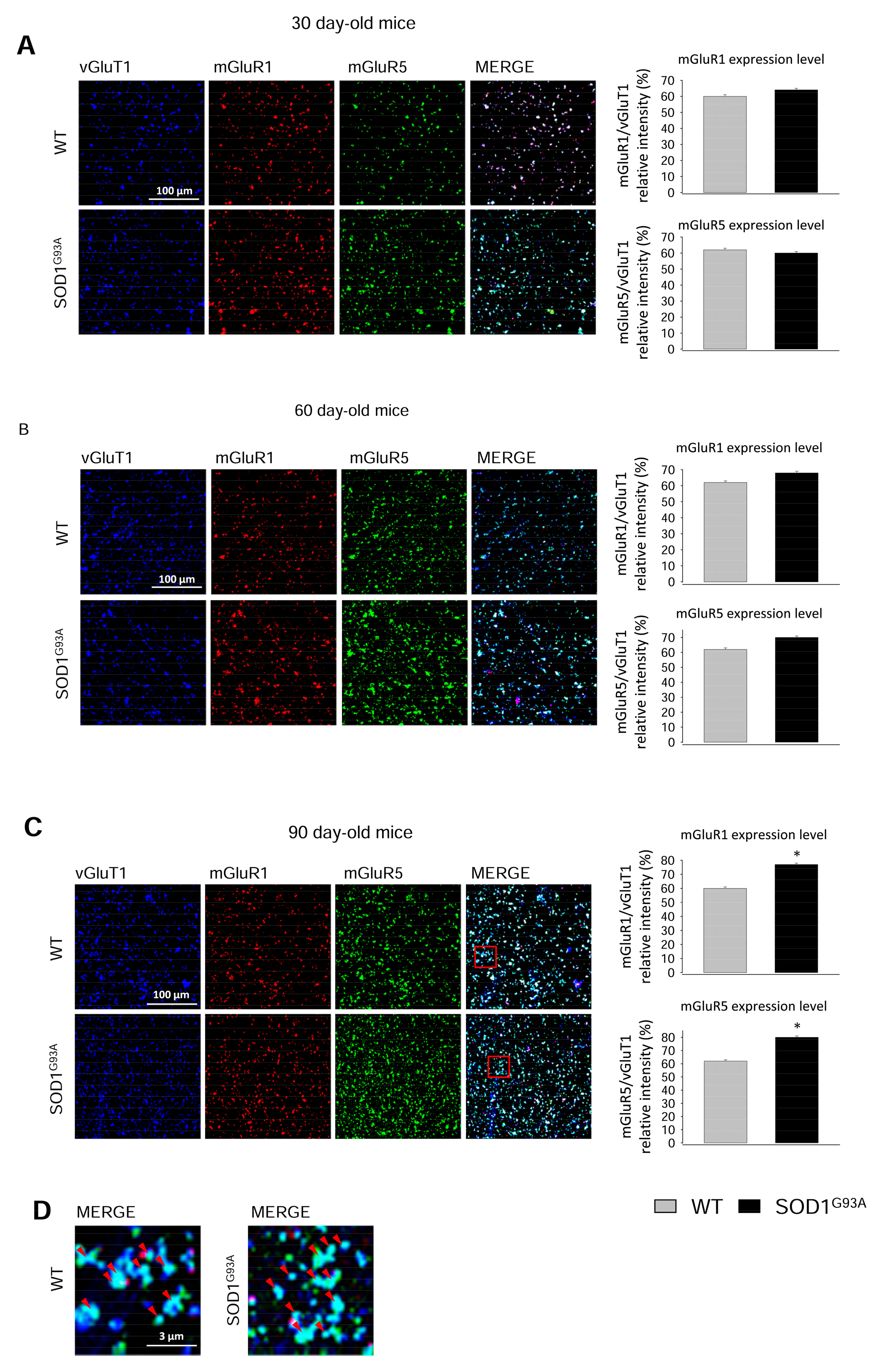
2.3. Expression of mGluR1 and mGluR5 in Spinal Cord Glutamatergic Synaptosomes during Disease Progression
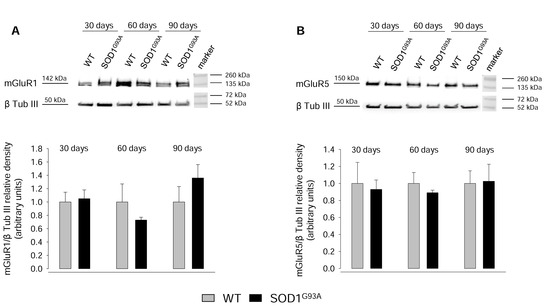
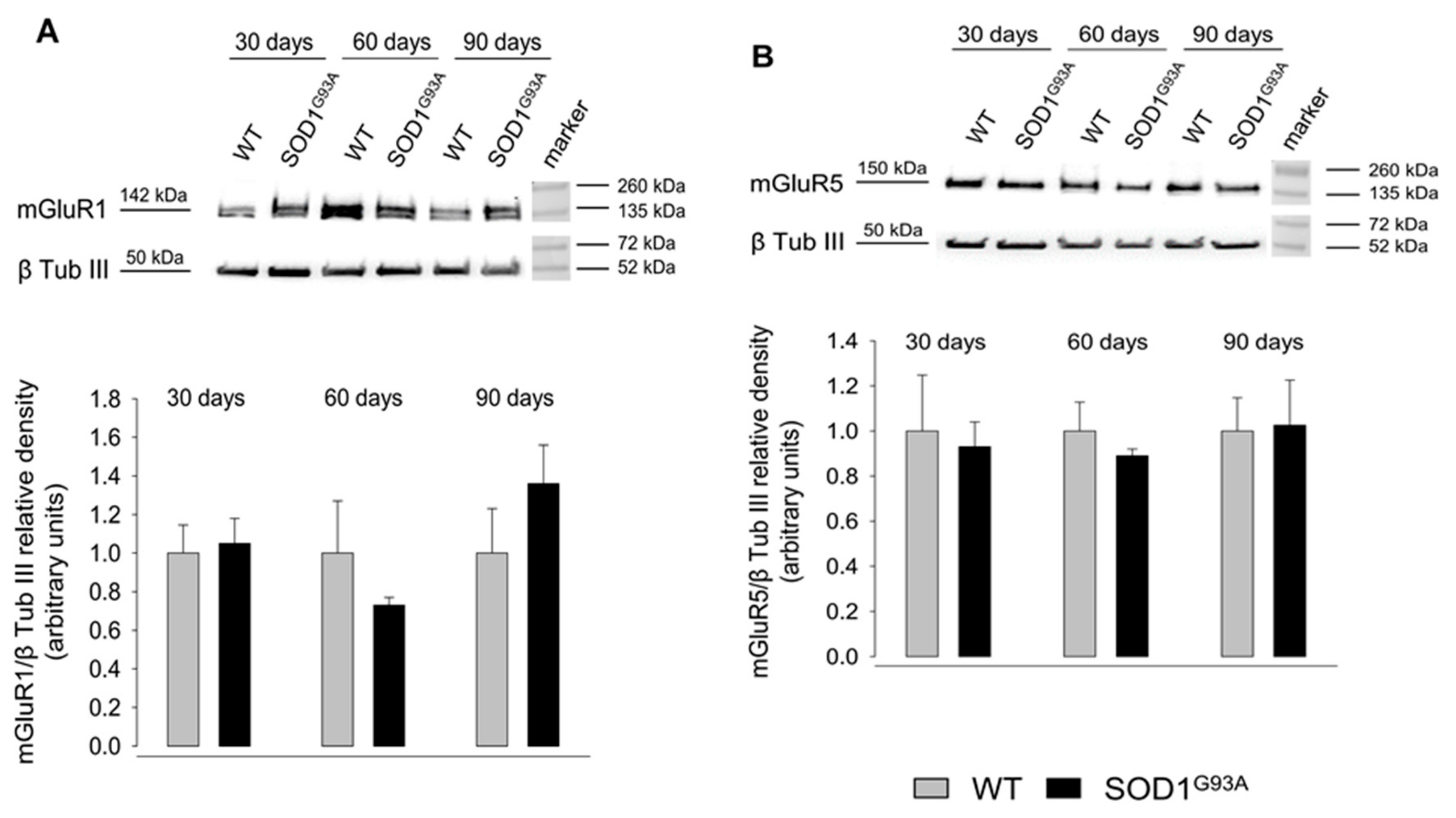
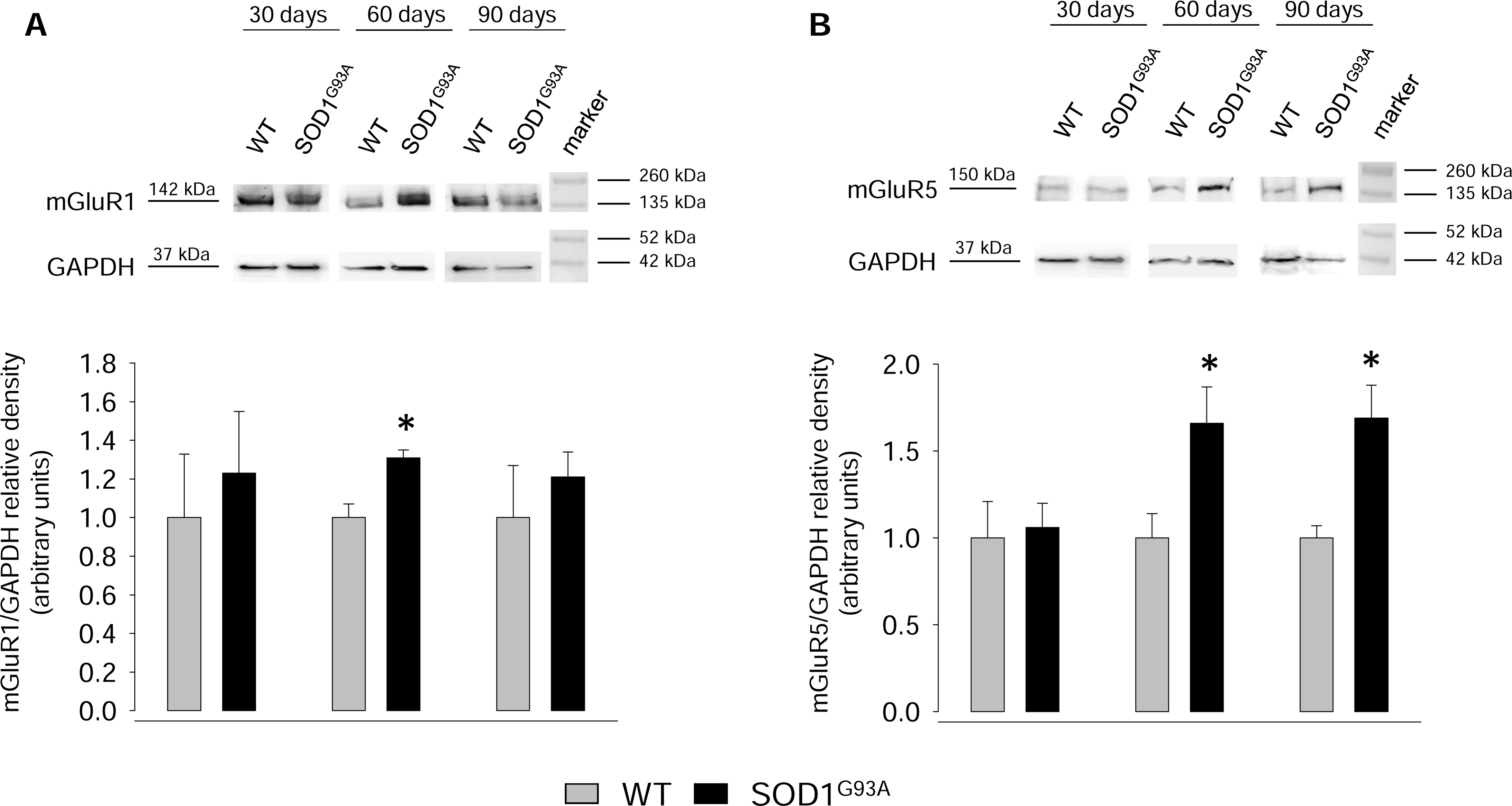
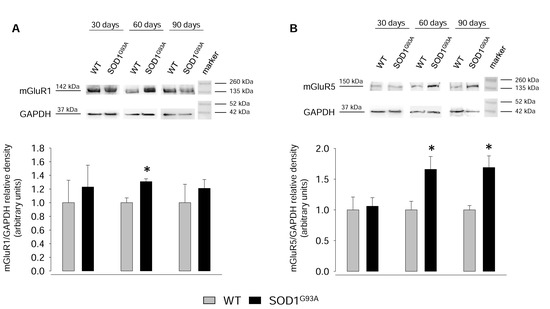
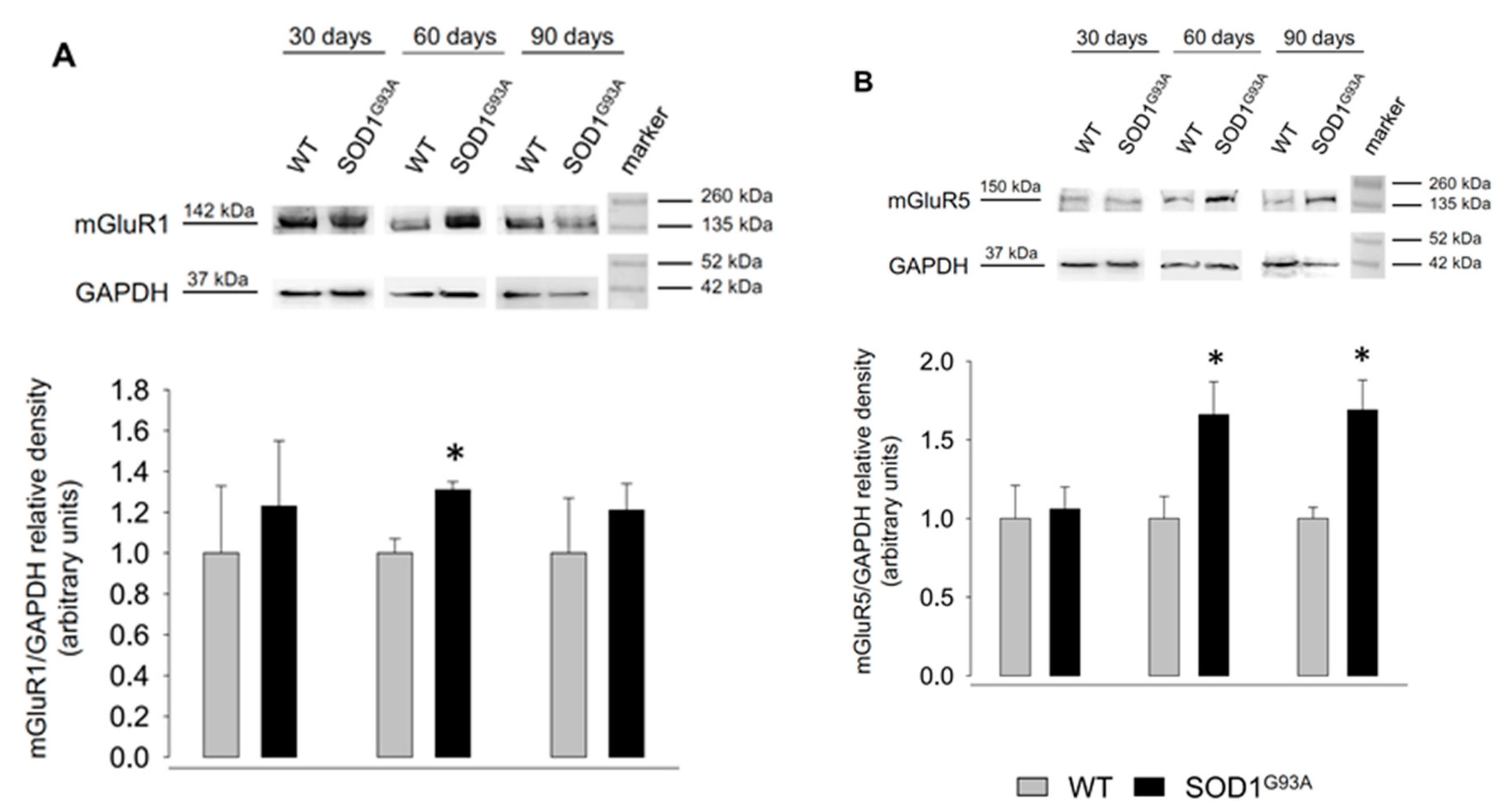
2.4. Expression of mGluR1 and mGluR5 in Spinal Cord Total Tissue during Disease Progression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Synaptosomes Purification
4.3. Release Experiments
4.4. Confocal Microscopy
4.5. Immunoblotting
4.6. Cytosolic Ca2+ Concentration
4.7. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, R.H., Jr. Amyotrophic lateral sclerosis: Recent insights from genetics and transgenic mice. Cell 1995, 80, 687–692. [Google Scholar] [CrossRef]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.; Bede, P.; Byrne, S.; Jordan, N.; Gallagher, L.; Wynne, B.; O’Brien, C.; Phukan, J.; Lynch, C.; Pender, N.; et al. Cognitive changes predict functional decline in ALS: A population-based longitudinal study. Neurology 2013, 80, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Le, W.D.; Xie, Y.Y.; Wang, X.P. Current Therapy of Drugs in Amyotrophic Lateral Sclerosis. Curr. Neuropharmacol. 2016, 14, 314–321. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Chiò, A.; Battistini, S.; Calvo, A.; Caponnetto, C.; Conforti, F.L.; Corbo, M.; Giannini, F.; Mandrioli, J.; Mora, G.; Sabatelli, M.; et al. Genetic counselling in ALS: Facts, uncertainties and clinical suggestions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 478–485. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Bruijn, L.I.; Wong, P.C.; Marszalek, J.R.; Vechio, J.D.; Lee, M.K.; Xu, X.S.; Borchelt, D.R.; Sisodia, S.S.; Price, D.L. Mechanisms of selective motorneuron death in transgenic mouse models of motorneuron disease. Neurology 1996, 47, S54–S61. [Google Scholar] [CrossRef]
- Morrison, B.M.; Morrison, J.H. Amyotrophic lateral sclerosis associated with mutations in superoxide dismutase: A putative mechanism of degeneration. Brain Res. Rev. 1999, 29, 121–135. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motorneuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motorneurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef]
- Shaw, P.J.; Forrest, V.; Ince, P.G.; Richardson, J.P.; Wastell, H.J. CSF and plasma amino acid levels in motorneuron disease: Elevation of CSF glutamate in a subset of patients. Neurodegeneration 1995, 4, 209–216. [Google Scholar] [CrossRef]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebro spinal fluid in amyotrophic lateral sclerosis: A reppraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-related changes in the cerebro spinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; Dykes-Hoberg, M.; Rothstein, J.D. Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann. Neurol. 2004, 55, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Foran, E.; Bogush, A.; Goffredo, M.; Roncaglia, P.; Gustincich, S.; Pasinelli, P.; Trotti, D. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia 2011, 59, 1719–1731. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Giribaldi, F.; Milanese, M.; Bonifacino, T.; Rossi, P.I.A.; Di Prisco, S.; Pittaluga, A.; Tacchetti, C.; Puliti, A.; Usai, C.; Bonanno, G. Group I metabotropic glutamate autoreceptors induce abnormal glutamate exocytosis in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology 2013, 66, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, L.; Paolucci, E.; Prisco, S.; Raiteri, M.; Bonanno, G. Activation of a glycine transporter on spinal cord neurons causes enhanced glutamate release in a mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2003, 138, 1021–1025. [Google Scholar] [CrossRef]
- Milanese, M.; Zappettini, S.; Jacchetti, E.; Bonifacino, T.; Cervetto, C.; Usai, C.; Bonanno, G. In vitro activation of GAT1 transporters expressed in spinal cord gliosomes stimulates glutamate release that is abnormally elevated in the SOD1/G93A(+) mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2010, 113, 489–501. [Google Scholar] [CrossRef]
- Milanese, M.; Bonifacino, T.; Fedele, E.; Rebosio, C.; Cattaneo, L.; Benfenati, F.; Usai, C.; Bonanno, G. Exocytosis regulates trafficking of GABA and glycine heterotransporters in spinal cord glutamatergic synapses: A mechanism for the excessive heterotransporter-induced release of glutamate in experimental amyotrophic lateral sclerosis. Neurobiol. Dis. 2015, 74, 314–324. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Zappettini, S.; Mercuri, N.B.; Raiteri, M.; Bonanno, G. Excessive and precocious glutamate release in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology 2004, 46, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Musazzi, L.; Milanese, M.; Seguini, M.; Marte, A.; Gallia, E.; Cattaneo, L.; Onofri, F.; Popoli, M.; Bonanno, G. Altered mechanisms underlying the abnormal glutamate release in amyotrophic lateral sclerosis at a pre-symptomatic stage of the disease. Neurobiol. Dis. 2016, 95, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, K.; Car, H. (S)-3,5-DHPG: A review. CNS Drug Rev. 2002, 8, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.; Battaglia, G.; Kingston, A.; O’Neill, M.J.; Catania, M.V.; Di Grezia, R.; Nicoletti, F. Neuroprotective activity of the potent and selective mGlu1a metabotropic glutamate receptor antagonist, (+)-2-methyl-4 carboxyphenylglycine (LY367385): Comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology 1999, 38, 199–207. [Google Scholar] [PubMed]
- Gasparini, F.; Lingenhöhl, K.; Stoehr, N.; Flor, P.J.; Heinrich, M.; Vranesic, I.; Biollaz, M.; Allgeier, H.; Heckendorn, R.; Urwyler, S.; et al. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 1999, 38, 1493–1503. [Google Scholar] [CrossRef]
- Pin, J.P.; Acher, F. The metabotropic glutamate receptors: Structure, activation mechanism and pharmacology. Curr. Drug Targets CNS Neurol. Disord. 2002, 1, 297–317. [Google Scholar] [CrossRef] [PubMed]
- Malgaroli, A.; Meldolesi, J. [Ca2+]i oscillations from internal stores sustain exocytic secretion from the chromaffin cells of the rat. FEBS Lett. 1991, 283, 169–172. [Google Scholar] [CrossRef]
- Bleasdale, J.E.; Thakur, N.R.; Gremban, R.S.; Bundy, G.L.; Fitzpatrick, F.A.; Smith, R.J.; Bunting, S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J. Pharmacol. Exp. 1990, 255, 756–768. [Google Scholar]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins (1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef]
- Biber, K.; Laurie, D.J.; Berthele, A.; Sommer, B.; Tölle, T.R.; Gebicke-Härter, P.J.; Van Calker, D.; Boddeke, H.W. Expression and signalling of group I metabotropic glutamate receptors in astrocytes and microglia. J. Neurochem. 1999, 72, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Luyt, K.; Varadi, A.; Molnar, E. Functional metabotropic glutamate receptors are expressed in oligodendrocyte progenitor cells. J. Neurochem. 2003, 84, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the work bench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Catania, M.V.; Geurts, J.; Yankaya, B.; Troost, D. Immuno-histochemical localization of group I and II metabotropic glutamate receptors in control and amyotrophic lateral sclerosis human spinal cord: Upregulation in reactive astrocytes. Neuroscience 2001, 105, 509–520. [Google Scholar] [CrossRef]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef]
- Anneser, J.M.; Ince, P.G.; Shaw, P.J.; Borasio, G.D. Differential expression of mGluR5 in human lumbosacral motoneurons. Neuroreport 2004, 15, 271–273. [Google Scholar] [CrossRef]
- Anneser, J.M.; Chahli, C.; Ince, P.G.; Borasio, G.D.; Shaw, P.J. Glial proliferation and metabotropic glutamate receptor expression in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2004, 63, 831–840. [Google Scholar] [CrossRef][Green Version]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef]
- Vergouts, M.; Doyen, P.J.; Peeters, M.; Opsomer, R.; Hermans, E. Constitutive downregulation protein kinase C epsilon in hSOD1G93A astrocytes influences mGluR5 signaling and the regulation of glutamate uptake. Glia 2018, 66, 749–761. [Google Scholar] [CrossRef]
- Musante, V.; Neri, E.; Feligioni, M.; Puliti, A.; Pedrazzi, M.; Conti, V.; Usai, C.; Diaspro, A.; Ravazzolo, R.; Henley, J.M.; et al. Presynaptic mGlu1 and mGlu5 autoreceptors facilitate glutamate exocytosis from mouse cortical nerve endings. Neuropharmacology 2008, 55, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Doumazane, E.; Scholler, P.; Zwier, J.M.; Trinquet, E.; Rondard, P.; Pin, J.P. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 2011, 25, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Beccaria, F.; Salamino, F.; Milanese, M.; Bonanno, G.; Pontremoli, S.; Melloni, E. Adaptive modifications in the calpain/calpastatin system in brain cells after persistent alteration in Ca2+ homeostasis. J. Biol. Chem. 2010, 285, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Milanese, M.; Bonifacino, T.; Bonanno, G.; Salamino, F.; Pontremoli, S.; Melloni, E. Role of calpain-1 in the early phase of experimental ALS. Arch. Biochem. Biophys. 2014, 562, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.; Turner, M.D. Emerging functions of the calpain superfamily of cysteine proteases in neuroendocrine secretory pathways. J. Neurochem. 2007, 103, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, G.; Raiteri, M. Release-regulating presynaptic heterocarriers. Prog. Neurobiol. 1994, 44, 451–462. [Google Scholar] [CrossRef]
- Di Prisco, S.; Merega, E.; Milanese, M.; Summa, M.; Casazza, S.; Raffaghello, L.; Pistoia, V.; Uccelli, A.; Pittaluga, A. CCL5-glutamate interaction in central nervous system: Early and acute presynaptic defects in EAE mice. Neuropharmacology 2013, 75, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Scheefhals, N.; MacGillavry, H.D. Functional organization of postsynaptic glutamate receptors. Mol. Cell. Neurosci. 2018, 91, 82–94. [Google Scholar] [CrossRef]
- D’Antoni, S.; Berretta, A.; Bonaccorso, C.M.; Bruno, V.; Aronica, E.; Nicoletti, F.; Catania, M.V. Metabotropic glutamate receptors in glial cells. Neurochem. Res. 2008, 33, 2436–2443. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Nonneman, A.; Robberecht, W.; Van Den Bosch, L. The role of oligodendroglial dysfunction in amyotrophic lateral sclerosis. Neurodegener Dis. Manag. 2014, 4, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Giribaldi, F.; Melone, M.; Bonifacino, T.; Musante, I.; Carminati, E.; Rossi, P.I.; Vergani, L.; Voci, A.; Conti, F.; et al. Knocking down metabotropic glutamate receptor 1 improves survival and disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 64, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Cattaneo, L.; Gallia, E.; Puliti, A.; Melone, M.; Provenzano, F.; Bossi, S.; Musante, I.; Usai, C.; Conti, F.; et al. In-vivo effects of knocking-down metabotropic glutamate receptor 5 in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neuropharmacology 2017, 123, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Provenzano, F.; Gallia, E.; Ravera, S.; Torazza, C.; Bossi, S.; Ferrando, S.; Puliti, A.; Van den Bosch, L.; Bonanno, G.; et al. In-vivo genetic ablation of metabotropic glutamate receptor type 5 slows down disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2019, 129, 79–92. [Google Scholar] [CrossRef]
- Pizzi, M.; Benarese, M.; Boroni, F.; Goffi, F.; Valerio, A.; Spano, P.F. Neuroprotection by metabotropic glutamate receptor agonists on kainate-induced degeneration of motor neurons in spinal cord slices from adult rat. Neuropharmacology 2000, 39, 903–910. [Google Scholar] [CrossRef]
- Anneser, J.M.; Chahli, C.; Borasio, G.D. Protective effect of metabotropic glutamate receptor inhibition on amyotrophic lateral sclerosis-cerebrospinal fluid toxicity in vitro. Neuroscience 2006, 141, 1879–1886. [Google Scholar] [CrossRef]
- D’Antoni, S.; Berretta, A.; Seminara, G.; Longone, P.; Giuffrida-Stella, A.M.; Battaglia, G.; Sortino, M.A.; Nicoletti, F.; Catania, M.V. A prolonged pharmacological blockade of type-5 metabotropic glutamate receptors protects cultured spinal cord motorneurons against excitotoxic death. Neurobiol. Dis. 2011, 42, 252–264. [Google Scholar] [CrossRef]
- Montana, M.C.; Cavallone, L.F.; Stubbert, K.K.; Stefanescu, A.D.; Kharasch, E.D.; Gereau, R.W. The metabotropic glutamate receptor subtype 5 antagonist fenobam is analgesic and has improved in vivo selectivity compared with the prototypical antagonist 2-methyl-6-(phenylethynyl)-pyridine. J. Pharmacol. Exp. Ther. 2009, 330, 834–843. [Google Scholar] [CrossRef]
- Lindemann, L.; Jaeschke, G.; Michalon, A.; Vieira, E.; Honer, M.; Spooren, W.; Porter, R.; Hartung, T.; Kolczewski, S.; Büttelmann, B.; et al. CTEP: A novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J. Pharmacol. Exp. Ther. 2011, 339, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Levenga, J.; Hayashi, S.; De Vrij, F.M.; Koekkoek, S.K.; Van der Linde, H.C.; Nieuwenhuizen, I.; Song, C.; Buijsen, R.A.; Pop, A.S.; Gomezmancilla, B.; et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis. 2011, 42, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Michalon, A.; Bruns, A.; Risterucci, C.; Honer, M.; Ballard, T.M.; Ozmen, L.; Jaeschke, G.; Wettstein, J.G.; Von Kienlin, M.; Künnecke, B.; et al. Chronic metabotropic glutamate receptor 5 inhibition corrects local alterations of brain activity and improves cognitive performance in fragile X mice. Biol. Psychiatry 2014, 75, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Peterlik, D.; Stangl, C.; Bauer, A.; Bludau, A.; Keller, J.; Grabski, D.; Killian, T.; Schmidt, D.; Zajicek, F.; Jaeschke, G.; et al. Blocking metabotropic glutamate receptor subtype 5 relieves maladaptive chronic stress consequences. Brain Behav. Immun. 2017, 59, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elrahman, K.S.; Hamilton, A.; Vasefi, M.; Ferguson, S.S.G. Autophagy is increased following either pharmacological or genetic silencing of mGluR5 signaling in Alzheimer’s disease mouse models. Mol. Brain 2018, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.H.; Jaeschke, G.; Spooren, W.; Ballard, T.M.; Büttelmann, B.; Kolczewski, S.; Peters, J.U.; Prinssen, E.; Wichmann, J.; Vieira, E.; et al. Fenobam: A clinically validated non-benzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J. Pharmacol. Exp. Ther. 2005, 315, 711–721. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hessl, D.; Coffey, S.; Hervey, C.; Schneider, A.; Yuhas, J.; Hutchison, J.; Snape, M.; Tranfaglia, M.; Nguyen, D.V.; et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 2009, 46, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, L.; Porter, R.H.; Scharf, S.H.; Kuennecke, B.; Bruns, A.; Von Kienlin, M.; Harrison, A.C.; Paehler, A.; Funk, C.; Gloge, A.; et al. Pharmacology of basimglurant (RO4917523, RG7090), a unique metabotropic glutamate receptor 5 negative allosteric modulator in clinical development for depression. J. Pharmacol. Exp. Ther. 2015, 353, 213–233. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Raiteri, L.; Stigliani, S.; Usai, C.; Diaspro, A.; Paluzzi, S.; Milanese, M.; Raiteri, M.; Bonanno, G. Functional expression of release-regulating glycine transporters GLYT1 on GABAergic neurons and GLYT2 on astrocytes in mouse spinal cord. Neurochem. Int. 2008, 52, 103–112. [Google Scholar] [CrossRef]
- Carney, K.E.; Milanese, M.; Van Nierop, P.; Li, K.W.; Oliet, S.H.R.; Smit, A.B.; Bonanno, G.; Verheijen, M.H.G. Proteomic analysis of gliosomes from mouse brain: Identification and investigation of glial membrane proteins. J. Proteome Res. 2014, 13, 5918–5927. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dyebinding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Fleck, M.W.; Barrionuevo, G.; Palmer, A.M. Synaptosomal and vesicular accumulation of L-glutamate, L-aspartate and D-aspartate. Neurochem. Int. 2001, 39, 217–225. [Google Scholar] [CrossRef]
- Raiteri, L.; Zappettini, S.; Milanese, M.; Fedele, E.; Raiteri, M.; Bonanno, G. Mechanisms of glutamate release elicited in rat cerebrocortical nerve endings by ‘pathologically’ elevated extraterminal K+ concentrations. J. Neurochem. 2007, 103, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, M.; Bonanno, G.; Marchi, M.; Maura, G. Is there a functional linkage between neurotransmitter uptake mechanisms and presynaptic receptors? J. Pharmacol. Exp. Ther. 1984, 231, 671–677. [Google Scholar] [PubMed]
- Bossi, S.; Musante, I.; Bonfiglio, T.; Bonifacino, T.; Emionite, L.; Cerminara, M.; Cervetto, C.; Marcoli, M.; Bonanno, G.; Ravazzolo, R.; et al. Genetic inactivation of mGlu5 receptor improves motor coordination in the Grm1crv4 mouse model of SCAR13 ataxia. Neurobiol. Dis. 2018, 109, 44–53. [Google Scholar] [CrossRef]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; Van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103, 857–862. [Google Scholar]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonifacino, T.; Rebosio, C.; Provenzano, F.; Torazza, C.; Balbi, M.; Milanese, M.; Raiteri, L.; Usai, C.; Fedele, E.; Bonanno, G. Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression. Int. J. Mol. Sci. 2019, 20, 4552. https://doi.org/10.3390/ijms20184552
Bonifacino T, Rebosio C, Provenzano F, Torazza C, Balbi M, Milanese M, Raiteri L, Usai C, Fedele E, Bonanno G. Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression. International Journal of Molecular Sciences. 2019; 20(18):4552. https://doi.org/10.3390/ijms20184552
Chicago/Turabian StyleBonifacino, Tiziana, Claudia Rebosio, Francesca Provenzano, Carola Torazza, Matilde Balbi, Marco Milanese, Luca Raiteri, Cesare Usai, Ernesto Fedele, and Giambattista Bonanno. 2019. "Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression" International Journal of Molecular Sciences 20, no. 18: 4552. https://doi.org/10.3390/ijms20184552
APA StyleBonifacino, T., Rebosio, C., Provenzano, F., Torazza, C., Balbi, M., Milanese, M., Raiteri, L., Usai, C., Fedele, E., & Bonanno, G. (2019). Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression. International Journal of Molecular Sciences, 20(18), 4552. https://doi.org/10.3390/ijms20184552