Identifying Methylation Pattern and Genes Associated with Breast Cancer Subtypes

 , ,
, ,  and
and 
Abstract
1. Introduction
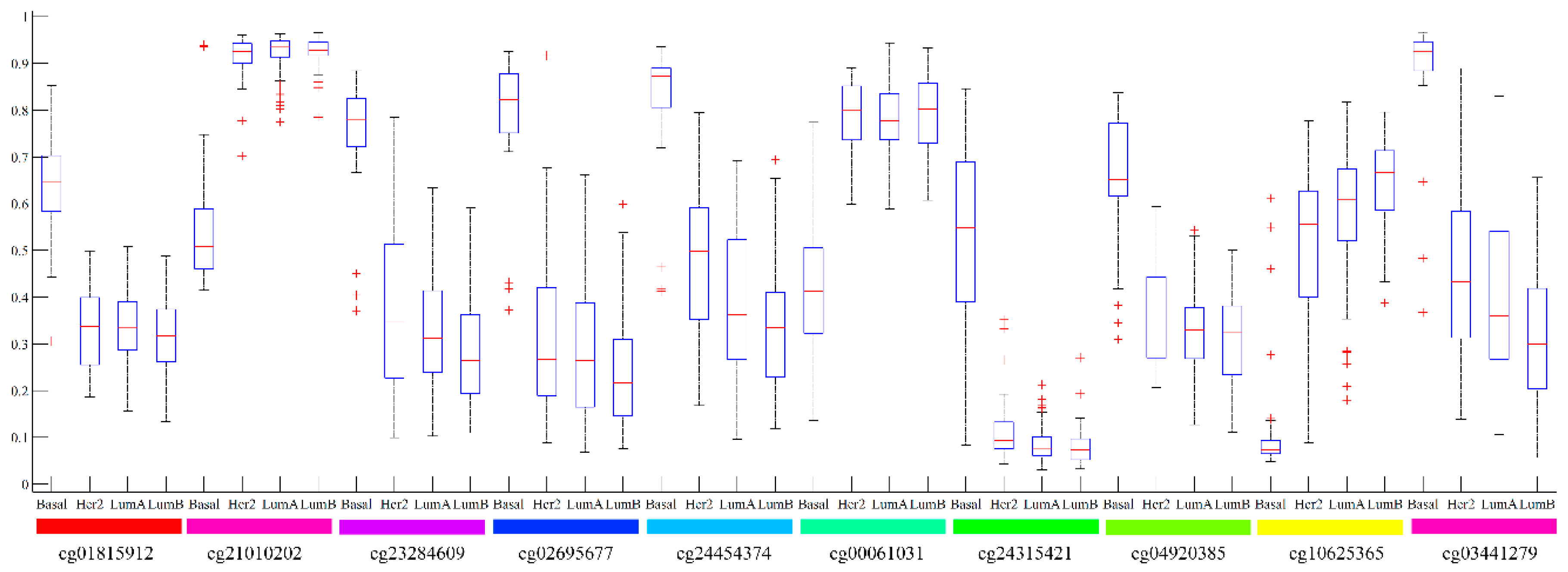
2. Results
3. Discussion
3.1. Analysis of Top Ranked Genes
3.2. Case Study on LumB
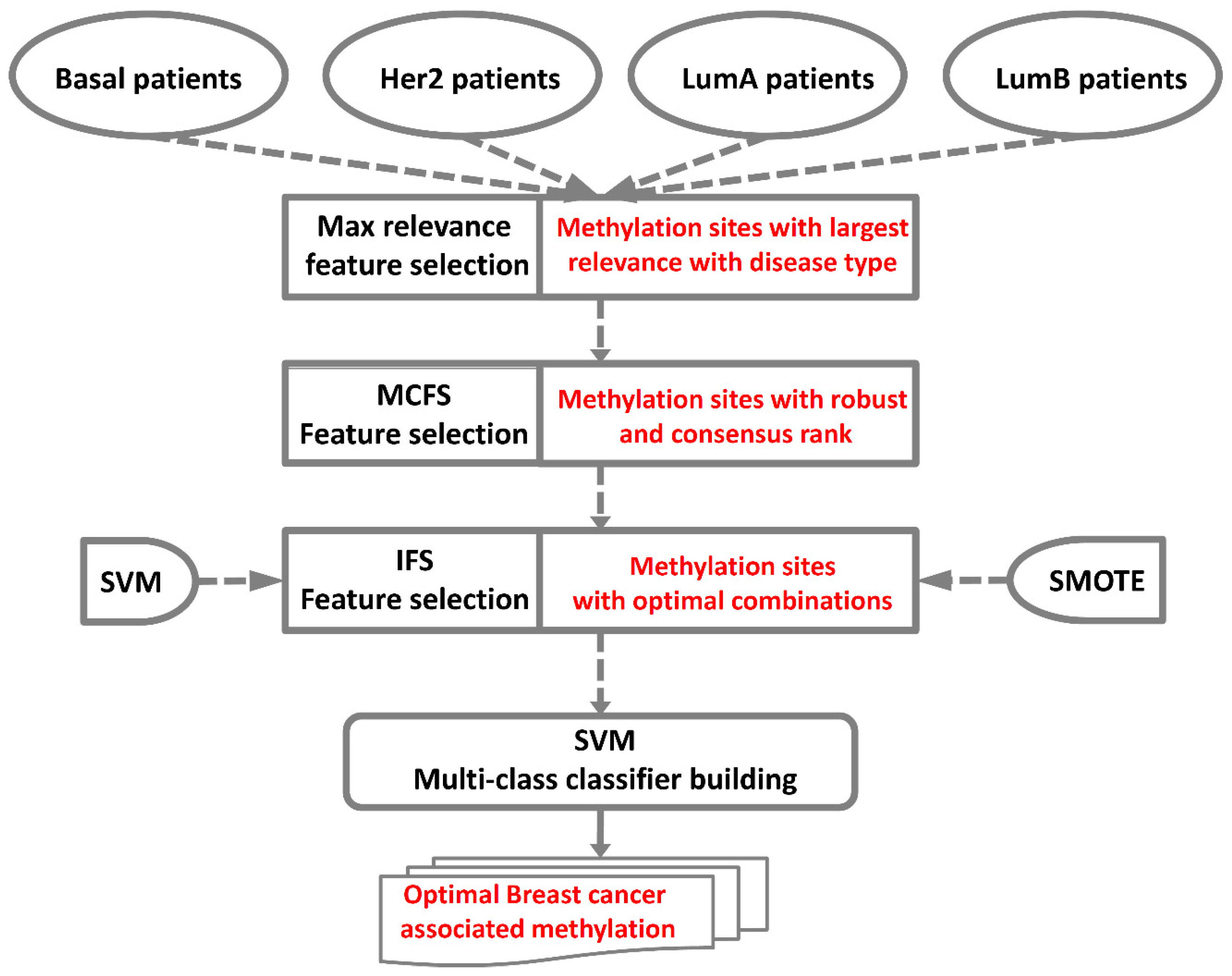
4. Materials and Methods
4.1. Datasets
4.2. Feature Selection
4.2.1. Maximum Relevance Score
4.2.2. Monte Carlo Feature Selection
4.2.3. Incremental Feature Selection
4.3. SVM
4.4. Synthetic Minority Over-Sampling Technique
4.5. Performance Measurement
4.6. Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Morris, M.; Woods, L.M.; Bhaskaran, K.; Rachet, B. Do pre-diagnosis primary care consultation patterns explain deprivation-specific differences in net survival among women with breast cancer? An examination of individually-linked data from the uk west midlands cancer registry, national screening programme and clinical practice research datalink. BMC Cancer 2017, 17, 155. [Google Scholar]
- Cedolini, C.; Bertozzi, S.; Londero, A.P.; Bernardi, S.; Seriau, L.; Concina, S.; Cattin, F.; Risaliti, A. Type of breast cancer diagnosis, screening, and survival. Clin. Breast Cancer 2014, 14, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, S.; Campbell, I.; Scott, N.; Shirley, R.; Lawrenson, R. Impact of mammographic screening on ethnic and socioeconomic inequities in breast cancer stage at diagnosis and survival in new zealand: A cohort study. BMC Public. Health 2015, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Richardson, A.; Frampton, C. Population attributable risks for modifiable lifestyle factors and breast cancer in new zealand women. Intern. Med. J. 2013, 43, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Anderson, A.S.; Clarke, R.B.; Duffy, S.W.; Evans, D.G.; Garcia-Closas, M.; Gescher, A.J.; Key, T.J.; Saxton, J.M.; Harvie, M.N. Risk determination and prevention of breast cancer. Breast Cancer Res. Bcr. 2014, 16, 446. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wen, W.; Zheng, Y.; Gao, Y.T.; Wu, C.; Bao, P.; Wang, C.; Gu, K.; Peng, P.; Gong, Y.; et al. Breast cancer incidence and mortality: Trends over 40 years among women in shanghai, china. Ann. Oncol. 2016, 27, 1129–1134. [Google Scholar] [CrossRef]
- Sung, H.; Ren, J.; Li, J.; Pfeiffer, R.M.; Wang, Y.; Guida, J.L.; Fang, Y.; Shi, J.; Zhang, K.; Li, N.; et al. Breast cancer risk factors and mammographic density among high-risk women in urban china. NPJ Breast Cancer 2018, 4, 3. [Google Scholar] [CrossRef]
- Nelson, H.D.; Pappas, M.; Zakher, B.; Mitchell, J.P.; Okinaka-Hu, L.; Fu, R. Risk assessment, genetic counseling, and genetic testing for brca-related cancer in women: A systematic review to update the u.S. Preventive services task force recommendation. Ann. Intern. Med. 2014, 160, 255–266. [Google Scholar] [CrossRef]
- Pan, X.; Hu, X.; Zhang, Y.-H.; Chen, L.; Zhu, L.; Wan, S.; Huang, T.; Cai, Y.-D. Identification of the copy number variant biomarkers for breast cancer subtypes. Mol. Genet. Genom. 2019, 294, 95–110. [Google Scholar] [CrossRef]
- Deb, S.; Wong, S.Q.; Li, J.; Do, H.; Weiss, J.; Byrne, D.; Chakrabarti, A.; Bosma, T.; kConFab, I.; Fellowes, A.; et al. Mutational profiling of familial male breast cancers reveals similarities with luminal a female breast cancer with rare tp53 mutations. Br. J. Cancer 2014, 111, 2351–2360. [Google Scholar] [CrossRef]
- Krishnamurti, U.; Silverman, J.F. Her2 in breast cancer: A review and update. Adv. Anat. Pathol. 2014, 21, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Gangi, A.; Cass, I.; Paik, D.; Barmparas, G.; Karlan, B.; Dang, C.; Li, A.; Walsh, C.; Rimel, B.J.; Amersi, F.F. Breast cancer following ovarian cancer in brca mutation carriers. JAMA Surg. 2014, 149, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Waldrep, A.R.; Avery, E.J.; Rose, F.F., Jr.; Midathada, M.V.; Tilford, J.A.; Kolberg, H.C.; Hutchins, M.R. Breast cancer subtype influences the accuracy of predicting pathologic response by imaging and clinical breast exam after neoadjuvant chemotherapy. Anticancer Res. 2016, 36, 5389–5395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buist, D.S.; Bosco, J.L.; Silliman, R.A.; Gold, H.T.; Field, T.; Yood, M.U.; Quinn, V.P.; Prout, M.; Lash, T.L.; Breast Cancer Outcomes in Older Women, I. Long-term surveillance mammography and mortality in older women with a history of early stage invasive breast cancer. Breast Cancer Res. Treat. 2013, 142, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Giannakeas, V.; Lubinski, J.; Gronwald, J.; Moller, P.; Armel, S.; Lynch, H.T.; Foulkes, W.D.; Kim-Sing, C.; Singer, C.; Neuhausen, S.L.; et al. Mammography screening and the risk of breast cancer in brca1 and brca2 mutation carriers: A prospective study. Breast Cancer Res. Treat. 2014, 147, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sana, M.; Malik, H.J. Current and emerging breast cancer biomarkers. J. Cancer Res. Ther. 2015, 11, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Weigel, M.T.; Dowsett, M. Current and emerging biomarkers in breast cancer: Prognosis and prediction. Endocr. -Relat. Cancer 2010, 17, R245–R262. [Google Scholar] [CrossRef]
- Wang, D.; Li, J.-R.; Zhang, Y.-H.; Chen, L.; Huang, T.; Cai, Y.-D. Identification of differentially expressed genes between original breast cancer and xenograft using machine learning algorithms. Genes 2018, 9, 155. [Google Scholar] [CrossRef]
- Cai, Y.D.; Zhang, Q.; Zhang, Y.H.; Chen, L.; Huang, T. Identification of genes associated with breast cancer metastasis to bone on a protein-protein interaction network with a shortest path algorithm. J. Proteome Res. 2017, 16, 1027–1038. [Google Scholar] [CrossRef]
- Li, X.C.; Liu, C.; Huang, T.; Zhong, Y. The occurrence of genetic alterations during the progression of breast carcinoma. BioMed Res. Int. 2016, 2016, 5237827. [Google Scholar] [CrossRef]
- Fleischer, T.; Tekpli, X.; Mathelier, A.; Wang, S.; Nebdal, D.; Dhakal, H.P.; Sahlberg, K.K.; Schlichting, E.; Oslo Breast Cancer Research, C.; Borresen-Dale, A.L.; et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat. Commun. 2017, 8, 1379. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, G.; Cava, C.; Castiglioni, I. Micrornas: New biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 2015, 5, 1122–1143. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Rueda, O.M.; Chin, S.F.; Curtis, C.; Dunning, M.J.; Aparicio, S.A.; Caldas, C. Genome-driven integrated classification of breast cancer validated in over 7500 samples. Genome Biol. 2014, 15, 431. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S. Molecular testing in breast cancer: A guide to current practices. Arch. Pathol. Lab. Med. 2016, 140, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer epigenetics: An introduction. Methods Mol. Biol. 2015, 1238, 3–25. [Google Scholar]
- Herceg, Z.; Ushijima, T. Introduction: Epigenetics and cancer. Adv. Genet. 2010, 70, 1–23. [Google Scholar] [PubMed]
- Zeleznik-Le, N.J. Introduction to progress and promise of epigenetics for diagnosis and therapy in cancer. Cancer Genet. 2015, 208, 165–166. [Google Scholar] [CrossRef]
- Santos, G.C., Jr.; da Silva, A.P.; Feldman, L.; Ventura, G.M.; Vassetzky, Y.; de Moura Gallo, C.V. Epigenetic modifications, chromatin distribution and tp53 transcription in a model of breast cancer progression. J. Cell. Biochem. 2015, 116, 533–541. [Google Scholar] [CrossRef]
- Stefansson, O.A.; Esteller, M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am. J. Pathol. 2013, 183, 1052–1063. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-generation machine learning for biological networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Kerschbaum, H.H.; Kainz, V.; Hermann, A. Sarcoplasmic calcium-binding protein-immunoreactive material in the central nervous system of the snail, helix pomatia. Brain Res. 1992, 597, 339–342. [Google Scholar] [CrossRef]
- Adorjan, P.; Distler, J.; Lipscher, E.; Model, F.; Muller, J.; Pelet, C.; Braun, A.; Florl, A.R.; Gutig, D.; Grabs, G.; et al. Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res. 2002, 30, e21. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pan, X.; Hu, X.; Zhang, Y.H.; Wang, S.; Huang, T.; Cai, Y.D. Gene expression differences among different msi statuses in colorectal cancer. Int. J. Cancer 2018, 143, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Shipp, M.A.; Ross, K.N.; Tamayo, P.; Weng, A.P.; Kutok, J.L.; Aguiar, R.C.; Gaasenbeek, M.; Angelo, M.; Reich, M.; Pinkus, G.S.; et al. Diffuse large b-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med. 2002, 8, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.H.; Qin, L.X.; Forgues, M.; He, P.; Kim, J.W.; Peng, A.C.; Simon, R.; Li, Y.; Robles, A.I.; Chen, Y.; et al. Predicting hepatitis b virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat. Med. 2003, 9, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Bernard, P.S.; Factor, R.E.; Kwan, M.L.; Habel, L.A.; Quesenberry, C.P., Jr.; Shakespear, K.; Weltzien, E.K.; Stijleman, I.J.; Davis, C.A.; et al. Intrinsic subtypes from pam50 gene expression assay in a population-based breast cancer cohort: Differences by age, race, and tumor characteristics. Cancer Epidemiol. Biomark. Prev. 2014, 23, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, L.; Guo, Z.-H.; Liu, T. Predicting drug side effects with compact integration of heterogeneous networks. Curr. Bioinform. 2019. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, L.; Lu, J. A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 2018, 306, 136–144. [Google Scholar] [CrossRef]
- De Summa, S.; Pinto, R.; Pilato, B.; Sambiasi, D.; Porcelli, L.; Guida, G.; Mattioli, E.; Paradiso, A.; Merla, G.; Micale, L.; et al. Expression of base excision repair key factors and mir17 in familial and sporadic breast cancer. Cell Death Dis. 2014, 5, e1076. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Evans, J.; Kim, K.; Chae, H.; Kim, S. Determining the effect of DNA methylation on gene expression in cancer cells. Methods Mol. Biol. 2014, 1101, 161–178. [Google Scholar] [PubMed]
- Ishizuka, T.; Rozehnal, V.; Fischer, T.; Kato, A.; Endo, S.; Yoshigae, Y.; Kurihara, A.; Izumi, T. Interindividual variability of carboxymethylenebutenolidase homolog, a novel olmesartan medoxomil hydrolase, in the human liver and intestine. Drug Metab. Dispos. 2013, 41, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lam, S.H.; Shen, Y.; Gong, Z. Genome-wide identification of molecular pathways and biomarkers in response to arsenic exposure in zebrafish liver. PLoS ONE 2013, 8, e68737. [Google Scholar] [CrossRef] [PubMed]
- Shashova, E.E.; Lyupina, Y.V.; Glushchenko, S.A.; Slonimskaya, E.M.; Savenkova, O.V.; Kulikov, A.M.; Gornostaev, N.G.; Kondakova, I.V.; Sharova, N.P. Proteasome functioning in breast cancer: Connection with clinical-pathological factors. PLoS ONE 2014, 9, e109933. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.S.; Gouvea, I.E.; Silva, M.C.; Castro, E.D.; de Paula, C.A.; Okamoto, D.; Oliveira, L.; Peres, G.B.; Ottaiano, T.; Facina, G.; et al. Cathepsin k induces platelet dysfunction and affects cell signaling in breast cancer—Molecularly distinct behavior of cathepsin k in breast cancer. BMC Cancer 2016, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Jin, T.; Geng, T.; Sun, T.; Li, X.; Dang, C.; Kang, L.; Chen, C.; Sun, J. Polymorphisms in esr1 and flj43663 are associated with breast cancer risk in the han population. Tumour Biol. 2014, 35, 2187–2190. [Google Scholar] [CrossRef]
- Li, L.; Zhang, G.Q.; Chen, H.; Zhao, Z.J.; Chen, H.Z.; Liu, H.; Wang, G.; Jia, Y.H.; Pan, S.H.; Kong, R.; et al. Plasma and tumor levels of linc-pint are diagnostic and prognostic biomarkers for pancreatic cancer. Oncotarget 2016, 7, 71773–71781. [Google Scholar] [CrossRef]
- Garitano-Trojaola, A.; Jose-Eneriz, E.S.; Ezponda, T.; Unfried, J.P.; Carrasco-Leon, A.; Razquin, N.; Barriocanal, M.; Vilas-Zornoza, A.; Sangro, B.; Segura, V.; et al. Deregulation of linc-pint in acute lymphoblastic leukemia is implicated in abnormal proliferation of leukemic cells. Oncotarget 2018, 9, 12842–12852. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, M.; Liu, C.; Zhang, X.; Li, W.; Cheng, H.; Zhu, J.; Zhang, M.; Chen, Z.; Zhang, B. Association study confirmed three breast cancer-specific molecular subtype-associated susceptibility loci in chinese han women. Oncologist 2017, 22, 890–894. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Tietgens, A.J.; Aponte, A.; Fredriksson, K.; Fanning, A.S.; Gucek, M.; Anderson, J.M. Biotin ligase tagging identifies proteins proximal to e-cadherin, including lipoma preferred partner, a regulator of epithelial cell-cell and cell-substrate adhesion. J. Cell Sci. 2014, 127, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Gregory Call, S.; Brereton, D.; Bullard, J.T.; Chung, J.Y.; Meacham, K.L.; Morrell, D.J.; Reeder, D.J.; Schuler, J.T.; Slade, A.D.; Hansen, M.D. A zyxin-nectin interaction facilitates zyxin localization to cell-cell adhesions. Biochem. Biophys. Res. Commun. 2011, 415, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.J.; Andrulis, I.L. Cell cycle regulated phosphorylation of limd1 in cell lines and expression in human breast cancers. Cancer Lett. 2008, 267, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.; Northey, J.J.; Brown, C.M.; Ursini-Siegel, J.; Siegel, P.M. A complex containing lpp and alpha-actinin mediates TGF β-induced migration and invasion of ERBB2-expressing breast cancer cells. J. Cell Sci. 2013, 126, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.; Stoletov, K.; Smith, H.W.; Common, J.; Muller, W.J.; Lewis, J.D.; Siegel, P.M. Lpp is a src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat. Commun. 2017, 8, 15059. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, L.; Reilly, P.T.; Shen, S.M.; He, P.; Zhu, X.N.; Li, C.X.; Wang, L.S.; Mak, T.W.; Chen, G.Q.; et al. Anp32b deficiency impairs proliferation and suppresses tumor progression by regulating akt phosphorylation. Cell Death Dis. 2016, 7, e2082. [Google Scholar] [CrossRef]
- Shen, S.M.; Yu, Y.; Wu, Y.L.; Cheng, J.K.; Wang, L.S.; Chen, G.Q. Downregulation of anp32b, a novel substrate of caspase-3, enhances caspase-3 activation and apoptosis induction in myeloid leukemic cells. Carcinogenesis 2010, 31, 419–426. [Google Scholar] [CrossRef]
- Leo, V.I.; Bunte, R.M.; Reilly, P.T. Balb/c-congenic anp32b-deficient mice reveal a modifying locus that determines viability. Exp. Anim. 2016, 65, 53–62. [Google Scholar] [CrossRef][Green Version]
- Cieply, B.; Park, J.W.; Nakauka-Ddamba, A.; Bebee, T.W.; Guo, Y.; Shang, X.; Lengner, C.J.; Xing, Y.; Carstens, R.P. Multiphasic and dynamic changes in alternative splicing during induction of pluripotency are coordinated by numerous rna-binding proteins. Cell Rep. 2016, 15, 247–255. [Google Scholar] [CrossRef]
- Lin, X.; Huang, X.; Uziel, T.; Hessler, P.; Albert, D.H.; Roberts-Rapp, L.A.; McDaniel, K.F.; Kati, W.M.; Shen, Y. Hexim1 as a robust pharmacodynamic marker for monitoring target engagement of bet family bromodomain inhibitors in tumors and surrogate tissues. Mol. Cancer Ther. 2017, 16, 388–396. [Google Scholar] [CrossRef]
- Zeng, H.; Qu, J.; Jin, N.; Xu, J.; Lin, C.; Chen, Y.; Yang, X.; He, X.; Tang, S.; Lan, X.; et al. Feedback activation of leukemia inhibitory factor receptor limits response to histone deacetylase inhibitors in breast cancer. Cancer Cell 2016, 30, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.Y.; Ha, S.Y.; Yu, E.J.; Lee, K.W.; Kim, J.H.; Kim, S.H. Znf282 (zinc finger protein 282), a novel e2f1 co-activator, promotes esophageal squamous cell carcinoma. Oncotarget 2014, 5, 12260–12272. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Lee, A.H.; Roberts, J.; Villena Salinas, N.M.; Hodi, Z.; Ellis, I.O.; Reis-Filho, J.S. Low-estrogen receptor-positive breast cancer: The impact of tissue sampling, choice of antibody, and molecular subtyping. J. Clin. Oncol. 2012, 30, 2929–2930. [Google Scholar] [CrossRef] [PubMed]
- Balleine, R.L.; Wilcken, N.R. High-risk estrogen-receptor-positive breast cancer: Identification and implications for therapy. Mol. Diagn. Ther. 2012, 16, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Parker, J.S.; Leung, S.; Voduc, D.; Ebbert, M.; Vickery, T.; Davies, S.R.; Snider, J.; Stijleman, I.J.; Reed, J.; et al. A comparison of pam50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin. Cancer Res. 2010, 16, 5222–5232. [Google Scholar] [CrossRef] [PubMed]
- Breusegem, S.Y.; Seaman, M.N.J. Genome-wide rnai screen reveals a role for multipass membrane proteins in endosome-to-golgi retrieval. Cell Rep. 2014, 9, 1931–1945. [Google Scholar] [CrossRef] [PubMed]
- Savci-Heijink, C.D.; Halfwerk, H.; Koster, J.; van de Vijver, M.J. A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res. Treat. 2016, 156, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted disruption of the bcl9/beta-catenin complex inhibits oncogenic wnt signaling. Sci. Transl. Med. 2012, 4, 148ra117. [Google Scholar] [CrossRef]
- Elsarraj, H.S.; Hong, Y.; Valdez, K.E.; Michaels, W.; Hook, M.; Smith, W.P.; Chien, J.; Herschkowitz, J.I.; Troester, M.A.; Beck, M.; et al. Expression profiling of in vivo ductal carcinoma in situ progression models identified b cell lymphoma-9 as a molecular driver of breast cancer invasion. Breast Cancer Res. 2015, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Toya, H.; Oyama, T.; Ohwada, S.; Togo, N.; Sakamoto, I.; Horiguchi, J.; Koibuchi, Y.; Adachi, S.; Jigami, T.; Nakajima, T.; et al. Immunohistochemical expression of the beta-catenin-interacting protein b9l is associated with histological high nuclear grade and immunohistochemical ERBB2/HER-2 expression in breast cancers. Cancer Sci. 2007, 98, 484–490. [Google Scholar] [CrossRef]
- Bastien, R.R.; Rodriguez-Lescure, A.; Ebbert, M.T.; Prat, A.; Munarriz, B.; Rowe, L.; Miller, P.; Ruiz-Borrego, M.; Anderson, D.; Lyons, B.; et al. Pam50 breast cancer subtyping by RT-qPCR and concordance with standard clinical molecular markers. BMC Med. Genom. 2012, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Ogi, S.; Fujita, H.; Kashihara, M.; Yamamoto, C.; Sonoda, K.; Okamoto, I.; Nakagawa, K.; Ohdo, S.; Tanaka, Y.; Kuwano, M.; et al. Sorting nexin 2-mediated membrane trafficking of c-met contributes to sensitivity of molecular-targeted drugs. Cancer Sci. 2013, 104, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Megias, D.; Bravo, J. Sorting nexin 6 interacts with breast cancer metastasis suppressor-1 and promotes transcriptional repression. J. Cell. Biochem. 2010, 111, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Bendris, N.; Williams, K.C.; Reis, C.R.; Welf, E.S.; Chen, P.H.; Lemmers, B.; Hahne, M.; Leong, H.S.; Schmid, S.L. Snx9 promotes metastasis by enhancing cancer cell invasion via differential regulation of rhogtpases. Mol. Biol. Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Lourenco, C.M.; Losfeld, M.E.; Buckingham, K.J.; Kircher, M.; Nickerson, D.A.; Shendure, J.; Bamshad, M.J.; University of Washington Center for Mendelian, G.; Freeze, H.H. Mutations in the translocon-associated protein complex subunit ssr3 cause a novel congenital disorder of glycosylation. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bano-Polo, M.; Martinez-Garay, C.A.; Grau, B.; Martinez-Gil, L.; Mingarro, I. Membrane insertion and topology of the translocon-associated protein (TRAP) gamma subunit. Biochim. Biophys. Acta Biomembr. 2017, 1859, 903–909. [Google Scholar] [CrossRef]
- Hadad, S.M.; Coates, P.; Jordan, L.B.; Dowling, R.J.; Chang, M.C.; Done, S.J.; Purdie, C.A.; Goodwin, P.J.; Stambolic, V.; Moulder-Thompson, S.; et al. Evidence for biological effects of metformin in operable breast cancer: Biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res. Treat. 2015, 150, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Brocker, C.; Orlicky, D.J.; Vasiliou, V. Molecular characterization, expression analysis, and role of aldh3b1 in the cellular protection against oxidative stress. Free Radic. Biol. Med. 2010, 49, 1432–1443. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Orlicky, D.J.; Vasiliou, V. Expression and initial characterization of human aldh3b1. Biochem. Biophys. Res. Commun. 2007, 356, 792–798. [Google Scholar] [CrossRef]
- Sladek, N.E. Transient induction of increased aldehyde dehydrogenase 3a1 levels in cultured human breast (adeno)carcinoma cell lines via 5’-upstream xenobiotic, and electrophile, responsive elements is, respectively, estrogen receptor-dependent and -independent. Chem. Biol. Interact. 2003, 143, 63–74. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhu, Y.; Liu, L.; Wang, H.; Jiang, S.; Hu, X.; Guo, J. Stk39 blockage by rna interference inhibits the proliferation and induces the apoptosis of renal cell carcinoma. Onco Targets Ther. 2018, 11, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Donner, K.M.; Hiltunen, T.P.; Hannila-Handelberg, T.; Suonsyrja, T.; Kontula, K. Stk39 variation predicts the ambulatory blood pressure response to losartan in hypertensive men. Hypertens. Res. 2012, 35, 107–114. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Astolfi, A.; Landuzzi, L.; Nicoletti, G.; De Giovanni, C.; Croci, S.; Palladini, A.; Ferrini, S.; Iezzi, M.; Musiani, P.; Cavallo, F.; et al. Gene expression analysis of immune-mediated arrest of tumorigenesis in a transgenic mouse model of her-2/neu-positive basal-like mammary carcinoma. Am. J. Pathol. 2005, 166, 1205–1216. [Google Scholar] [CrossRef]
- Balatoni, C.E.; Dawson, D.W.; Suh, J.; Sherman, M.H.; Sanders, G.; Hong, J.S.; Frank, M.J.; Malone, C.S.; Said, J.W.; Teitell, M.A. Epigenetic silencing of STK39 in b-cell lymphoma inhibits apoptosis from genotoxic stress. Am. J. Pathol. 2009, 175, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.P. Cux1 mediates tumour cell survival: Implications for future therapies? Gut 2010, 59, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; Sebastian-Serrano, A.; Beccari, L.; Calcagnotto, M.E.; Cisneros, E.; Kim, S.; Dopazo, A.; Alvarez-Dolado, M.; Redondo, J.M.; Bovolenta, P.; et al. Cux1 and cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron 2010, 66, 523–535. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Z.; Yao, Y.; Dai, J.; Zhou, D.; Wang, L.; Zhang, Q.Q. Dipalmitoylphosphatidic acid inhibits breast cancer growth by suppressing angiogenesis via inhibition of the CUX1/FGF1/HGF signalling pathway. J. Cell. Mol. Med. 2018, 22, 4760–4770. [Google Scholar] [CrossRef]
- Hulea, L.; Nepveu, A. Cux1 transcription factors: From biochemical activities and cell-based assays to mouse models and human diseases. Gene 2012, 497, 18–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, S.; Yuan, F.; Zhang, K.; Fan, Y.; Zheng, S.; Gao, Z.; Zhao, J.; Mu, T.; Zhao, S.; et al. Mir-135 promotes proliferation and stemness of oesophageal squamous cell carcinoma by targeting rerg. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1210–1219. [Google Scholar] [CrossRef]
- Habashy, H.O.; Powe, D.G.; Glaab, E.; Ball, G.; Spiteri, I.; Krasnogor, N.; Garibaldi, J.M.; Rakha, E.A.; Green, A.R.; Caldas, C.; et al. Rerg (ras-like, oestrogen-regulated, growth-inhibitor) expression in breast cancer: A marker of er-positive luminal-like subtype. Breast Cancer Res. Treat. 2011, 128, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Finlin, B.S.; Gau, C.L.; Murphy, G.A.; Shao, H.; Kimel, T.; Seitz, R.S.; Chiu, Y.F.; Botstein, D.; Brown, P.O.; Der, C.J.; et al. Rerg is a novel ras-related, estrogen-regulated and growth-inhibitory gene in breast cancer. J. Biol. Chem. 2001, 276, 42259–42267. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, L.; Zhang, Y.H.; Liu, M.; Chen, L.; Huang, T.; Cai, Y.D. Identification of synthetic lethality based on a functional network by using machine learning algorithms. J. Cell. Biochem. 2019, 120, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pan, X.; Zhang, Y.-H.; Liu, M.; Huang, T.; Cai, Y.-D. Classification of widely and rarely expressed genes with recurrent neural network. Comput. Struct. Biotechnol. J. 2019, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.M.; Huang, T.; Wang, R.F. Cross talk of chromosome instability, cpg island methylator phenotype and mismatch repair in colorectal cancer. Oncol. Lett. 2018, 16, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, T. Predicting and analyzing early wake-up associated gene expressions by integrating gwas and eqtl studies. Biochim. Et Biophys. Acta Mol. Basis Dis. 2018, 1864, 2241–2246. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.H.; Huang, G.; Pan, X.; Wang, S.; Huang, T.; Cai, Y.D. Discriminating cirrnas from other lncrnas using a hierarchical extreme learning machine (H-ELM) algorithm with feature selection. Mol. Genet. Genom. 2018, 293, 137–149. [Google Scholar] [CrossRef]
- Chen, L.; Wang, S.; Zhang, Y.H.; Wei, L.; Xu, X.; Huang, T.; Cai, Y.D. Prediction of nitrated tyrosine residues in protein sequences by extreme learning machine and feature selection methods. Comb Chem High Throughput Screen 2018, 21, 393–402. [Google Scholar] [CrossRef]
- Cai, L.; Huang, T.; Su, J.; Zhang, X.; Chen, W.; Zhang, F.; He, L.; Chou, K.-C. Implications of newly identified brain EQTL genes and their interactors in schizophrenia. Mol. Ther.-Nucleic Acids 2018, 12, 433–442. [Google Scholar] [CrossRef]
- Wang, S.-B.; Huang, T.J.M.B.R. The early detection of asthma based on blood gene expression. Mol. Biol. Rep. 2019, 46, 217–223. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S.; Pan, X.; Hu, X.; Zhang, Y.H.; Yuan, F.; Huang, T.; Cai, Y.D. Hiv infection alters the human epigenetic landscape. Gene Ther. 2019, 26, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, X.; Dong, W.; Yan, J.; Liu, Q.; Zha, H. Joint active learning with feature selection via cur matrix decomposition. IEEE Trans. Pattern Anal. Mach. Intell. 2018, 41, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pan, X.; Zhang, Y.-H.; Kong, X.; Huang, T.; Cai, Y.-D. Tissue differences revealed by gene expression profiles of various cell lines. J. Cell. Biochem. 2019, 120, 7068–7081. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Hu, X.; Zhang, Y.H.; Feng, K.; Wang, S.P.; Chen, L.; Huang, T.; Cai, Y.D. Identifying patients with atrioventricular septal defect in down syndrome populations by using self-normalizing neural networks and feature selection. Genes 2018, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Zhang, Y.H.; Feng, K.; Wang, S.; Zhang, Y.; Huang, T.; Kong, X.; Cai, Y.D. Identification of gene expression signatures across different types of neural stem cells with the monte-carlo feature selection method. J. Cell. Biochem. 2018, 119, 3394–3403. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, L.; Feng, K.-Y.; Hu, X.-H.; Zhang, Y.-H.; Kong, X.-Y.; Huang, T.; Cai, Y.-D. Analysis of expression pattern of snornas in different cancer types with machine learning algorithms. Int. J. Mol. Sci. 2019, 20, 2185. [Google Scholar] [CrossRef]
- Chen, L.; Pan, X.; Zhang, Y.-H.; Hu, X.; Feng, K.; Huang, T.; Cai, Y.-D. Primary tumor site specificity is preserved in patient-derived tumor xenograft models. Front. Genet. 2019. [Google Scholar] [CrossRef]
- Chen, L.; Pan, X.; Zhang, Y.-H.; Huang, T.; Cai, Y.-D. Analysis of gene expression differences between different pancreatic cells. ACS Omega 2019, 4, 6421–6435. [Google Scholar] [CrossRef]
- Li, J.; Lan, C.-N.; Kong, Y.; Feng, S.-S.; Huang, T. Identification and analysis of blood gene expression signature for osteoarthritis with advanced feature selection methods. Front. Genet. 2018, 9, 246. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Zhang, Y.-H.; Kong, X.; Huang, T.; Cai, Y.-D. A computational method for classifying different human tissues with quantitatively tissue-specific expressed genes. Genes 2018, 9, 449. [Google Scholar] [CrossRef]
- Cui, H.; Chen, L. A binary classifier for the prediction of ec numbers of enzymes. Curr. Proteom. 2019, 16, 381–389. [Google Scholar] [CrossRef]
- Witten, I.H.; Frank, E. Data Mining: Practical Machine Learning Tools and Techniques; Morgan Kaufmann: San Francisco, CA, USA, 2005. [Google Scholar]
- Platt, J. Fast Training of Support Vector Machines Using Sequential Minimal Optimization; MIT Press: Cambridge, UK, 1998. [Google Scholar]
- Keerthi, S.S.; Shevade, S.K.; Bhattacharyya, C.; Murthy, K.R.K. Improvements to platt’s smo algorithm for svm classifier design. Neural. Comput. 2001, 13, 637–649. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In International Joint Conference on Artificial Intelligence; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA; Lawrence Erlbaum Associates: Mahwah, NJ, USA, 1995; pp. 1137–1145. [Google Scholar]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Li, J.; Xing, Z.-H.; Yang, J.; Huang, T.; Cai, Y.-D. Identify key sequence features to improve crispr sgrna efficacy. IEEE Access 2017, 5, 26582–26590. [Google Scholar] [CrossRef]
- Che, J.; Chen, L.; Guo, Z.-H.; Wang, S.; Aorigele. Drug target group prediction with multiple drug networks. Comb. Chem. High Throughput Screen. 2019. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Et Biophys. Acta (BBA)-Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Chen, L.; Chu, C.; Zhang, Y.-H.; Zheng, M.-Y.; Zhu, L.; Kong, X.; Huang, T. Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 2017, 12, 526–534. [Google Scholar] [CrossRef]
- Gorodkin, J. Comparing two k-category assignments by a k-category correlation coefficient. Comput. Biol. Chem. 2004, 28, 367–374. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | Sensitivity/Specificity | SVM | RF |
|---|---|---|---|
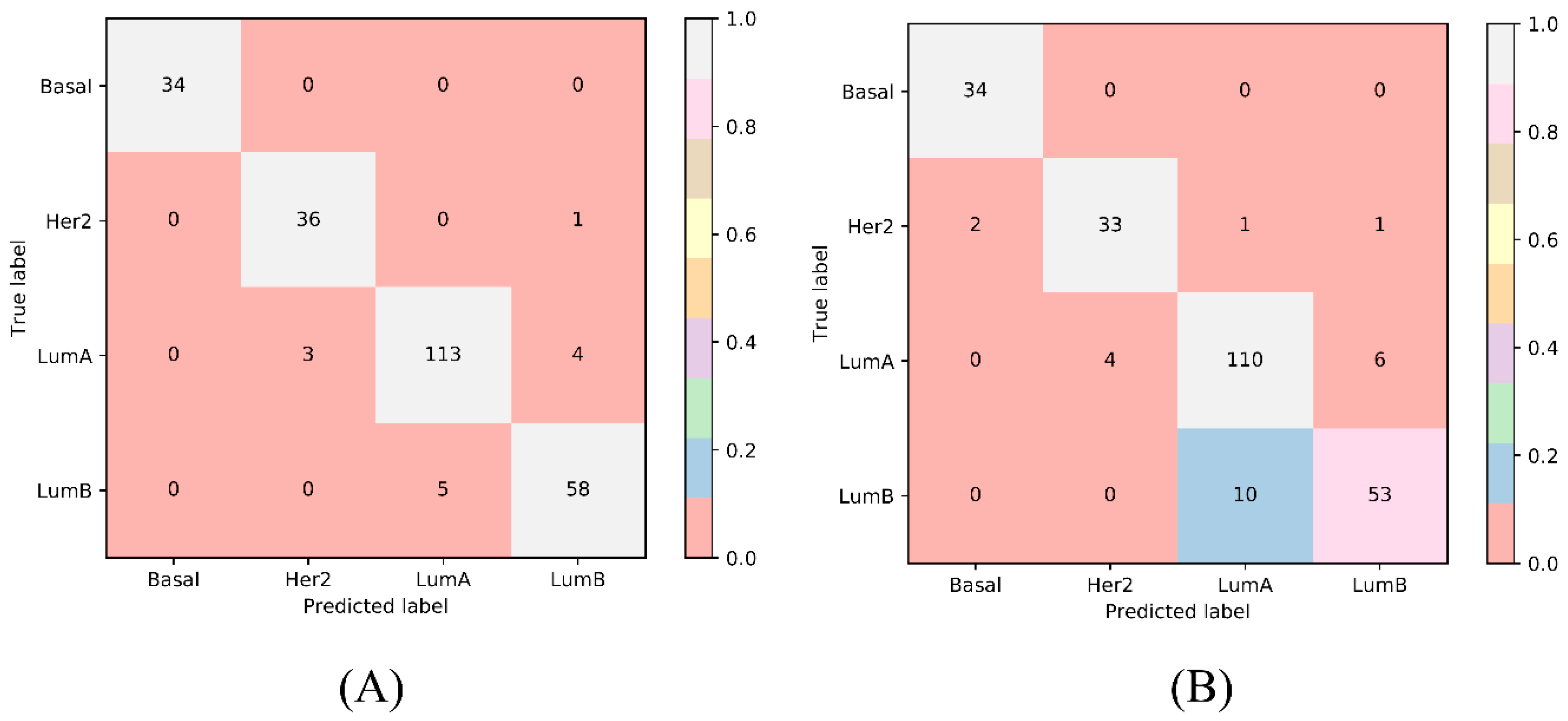
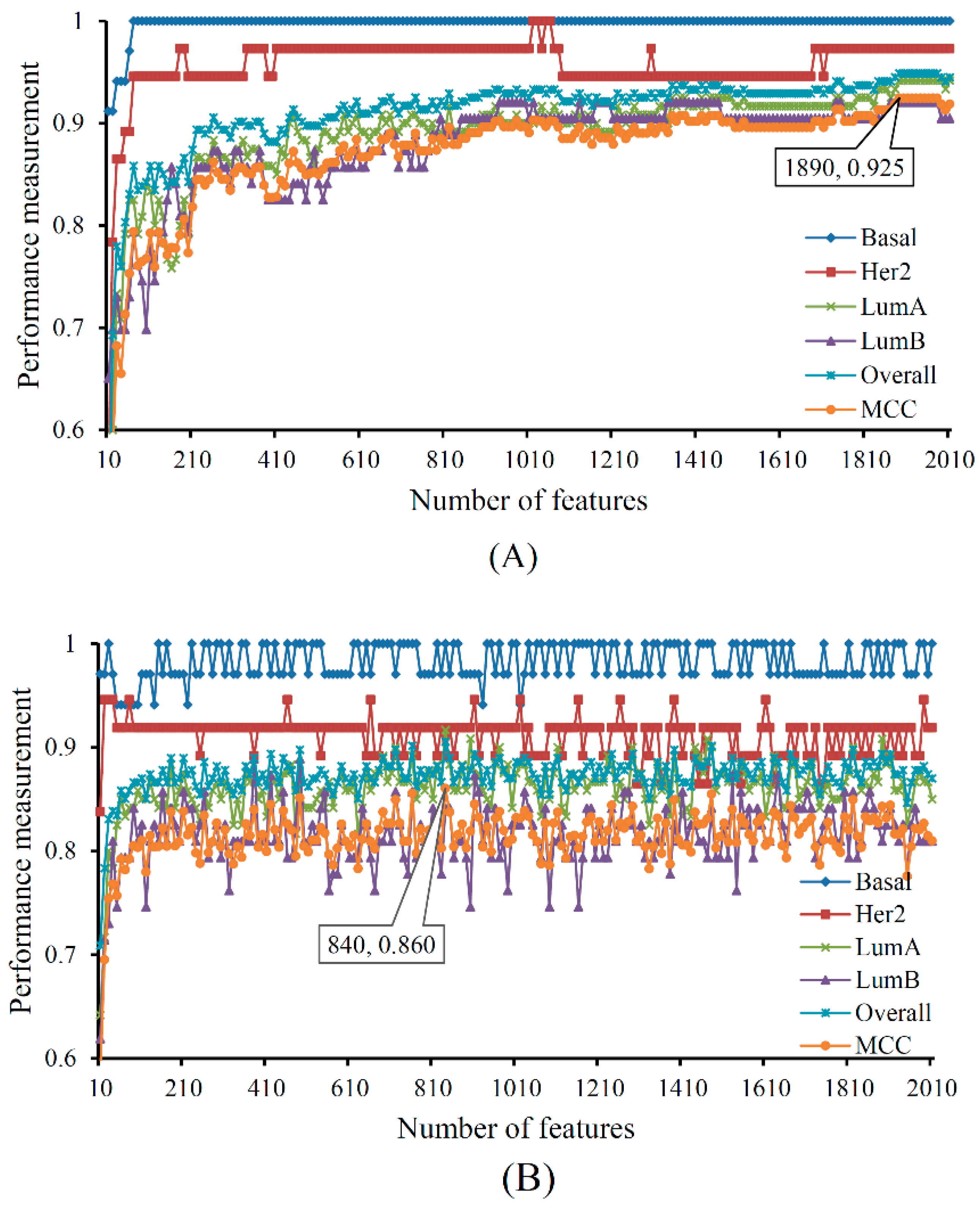
| Number of optimum features | / | 1890 | 840 |
| Matthews correlation coefficient (MCC) | / | 0.925 | 0.860 |
| Overall accuracy | / | 0.949 | 0.906 |
| Basal | Sensitivity | 1.000 | 1.000 |
| Specificity | 1.000 | 0.991 | |
| Her2 | Sensitivity | 0.973 | 0.892 |
| Specificity | 0.986 | 0.982 | |
| LumA | Sensitivity | 0.942 | 0.917 |
| Specificity | 0.963 | 0.918 | |
| LumB | Sensitivity | 0.921 | 0.841 |
| Specificity | 0.974 | 0.963 |
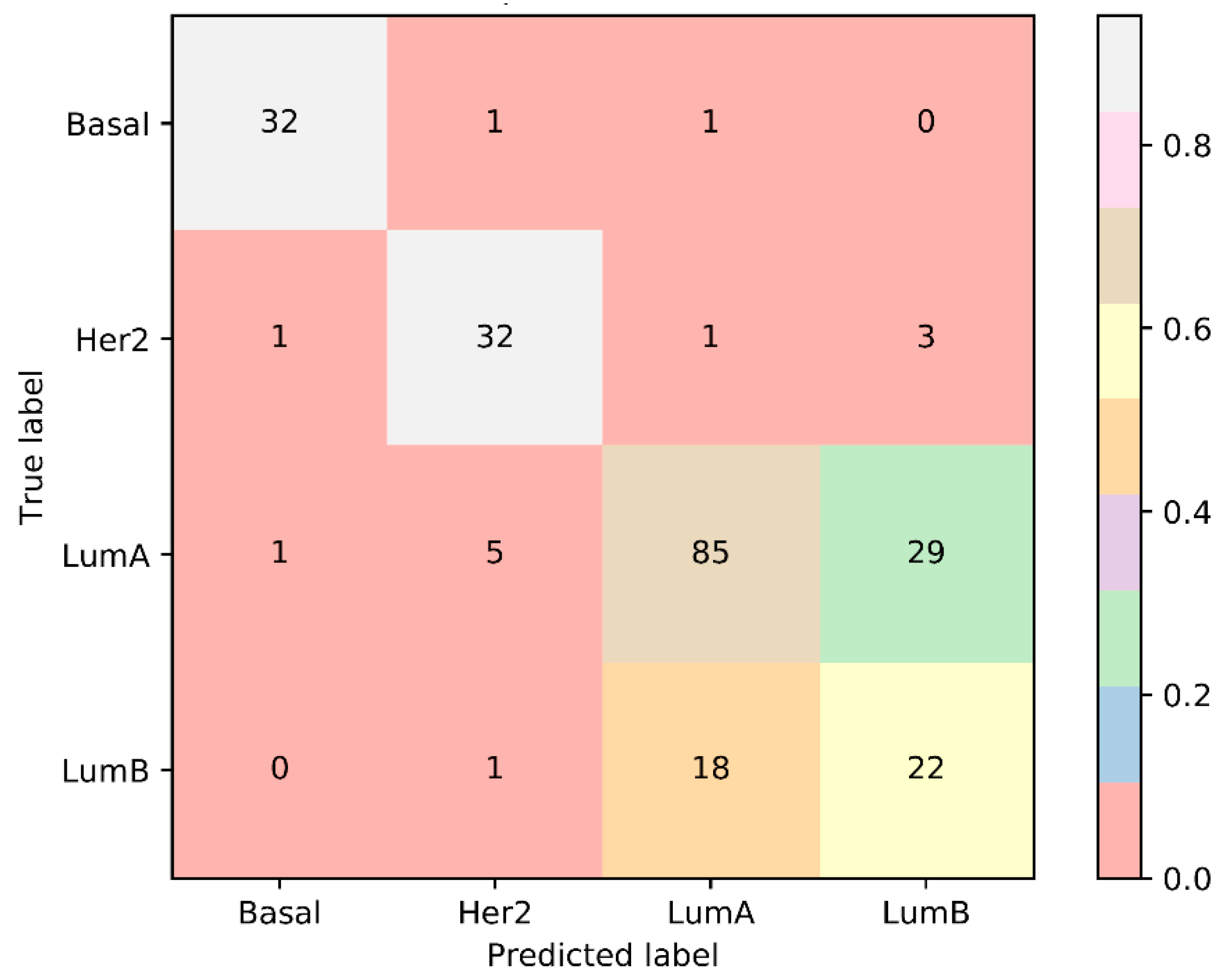
| Terms | Sensitivity/Specificity | SVM | RF |
|---|---|---|---|
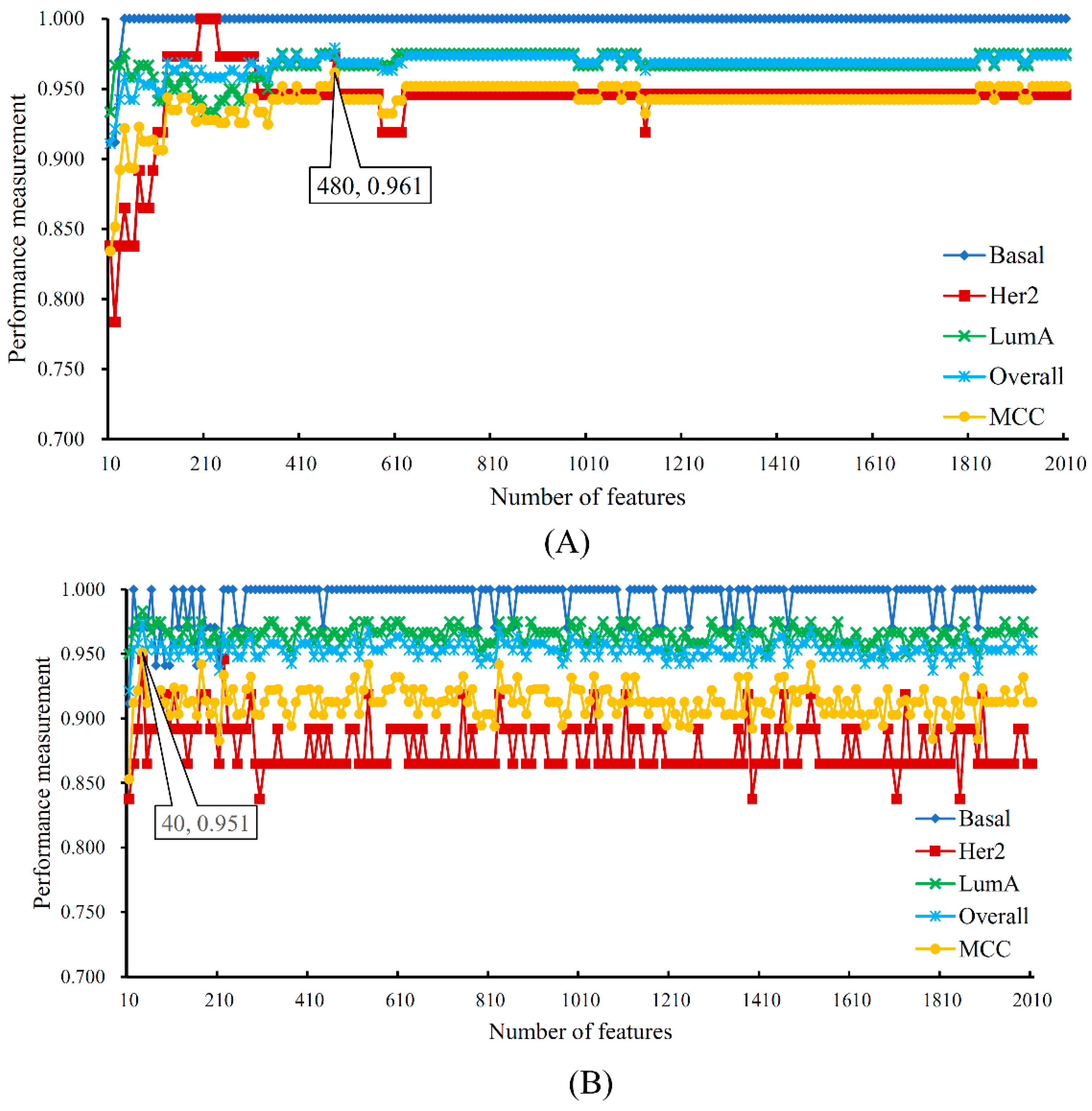
| Number of optimum features | / | 480 | 40 |
| MCC | / | 0.961 | 0.951 |
| Overall accuracy | / | 0.979 | 0.974 |
| Basal | Sensitivity | 1.000 | 0.971 |
| Specificity | 1.000 | 0.994 | |
| Her2 | Sensitivity | 0.973 | 0.946 |
| Specificity | 0.981 | 0.987 | |
| LumA | Sensitivity | 0.975 | 0.983 |
| Specificity | 0.986 | 0.972 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Zeng, T.; Pan, X.; Zhang, Y.-H.; Huang, T.; Cai, Y.-D. Identifying Methylation Pattern and Genes Associated with Breast Cancer Subtypes. Int. J. Mol. Sci. 2019, 20, 4269. https://doi.org/10.3390/ijms20174269
Chen L, Zeng T, Pan X, Zhang Y-H, Huang T, Cai Y-D. Identifying Methylation Pattern and Genes Associated with Breast Cancer Subtypes. International Journal of Molecular Sciences. 2019; 20(17):4269. https://doi.org/10.3390/ijms20174269
Chicago/Turabian StyleChen, Lei, Tao Zeng, Xiaoyong Pan, Yu-Hang Zhang, Tao Huang, and Yu-Dong Cai. 2019. "Identifying Methylation Pattern and Genes Associated with Breast Cancer Subtypes" International Journal of Molecular Sciences 20, no. 17: 4269. https://doi.org/10.3390/ijms20174269
APA StyleChen, L., Zeng, T., Pan, X., Zhang, Y.-H., Huang, T., & Cai, Y.-D. (2019). Identifying Methylation Pattern and Genes Associated with Breast Cancer Subtypes. International Journal of Molecular Sciences, 20(17), 4269. https://doi.org/10.3390/ijms20174269