Bone Diseases in Patients with Chronic Liver Disease


Abstract
1. Introduction
2. Definitions of Related Terms
3. Guidelines for Bone Diseases in Patients with Chronic Liver Disease
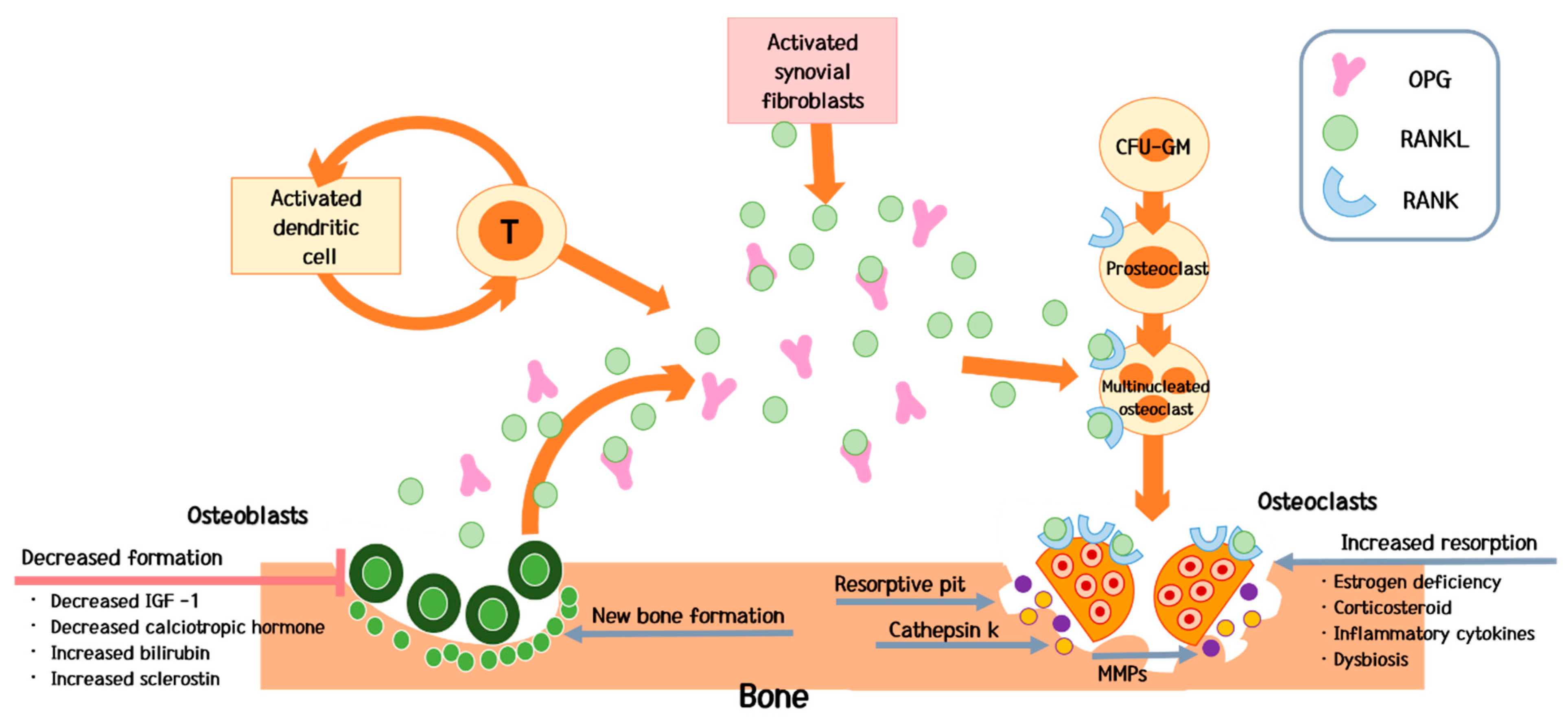
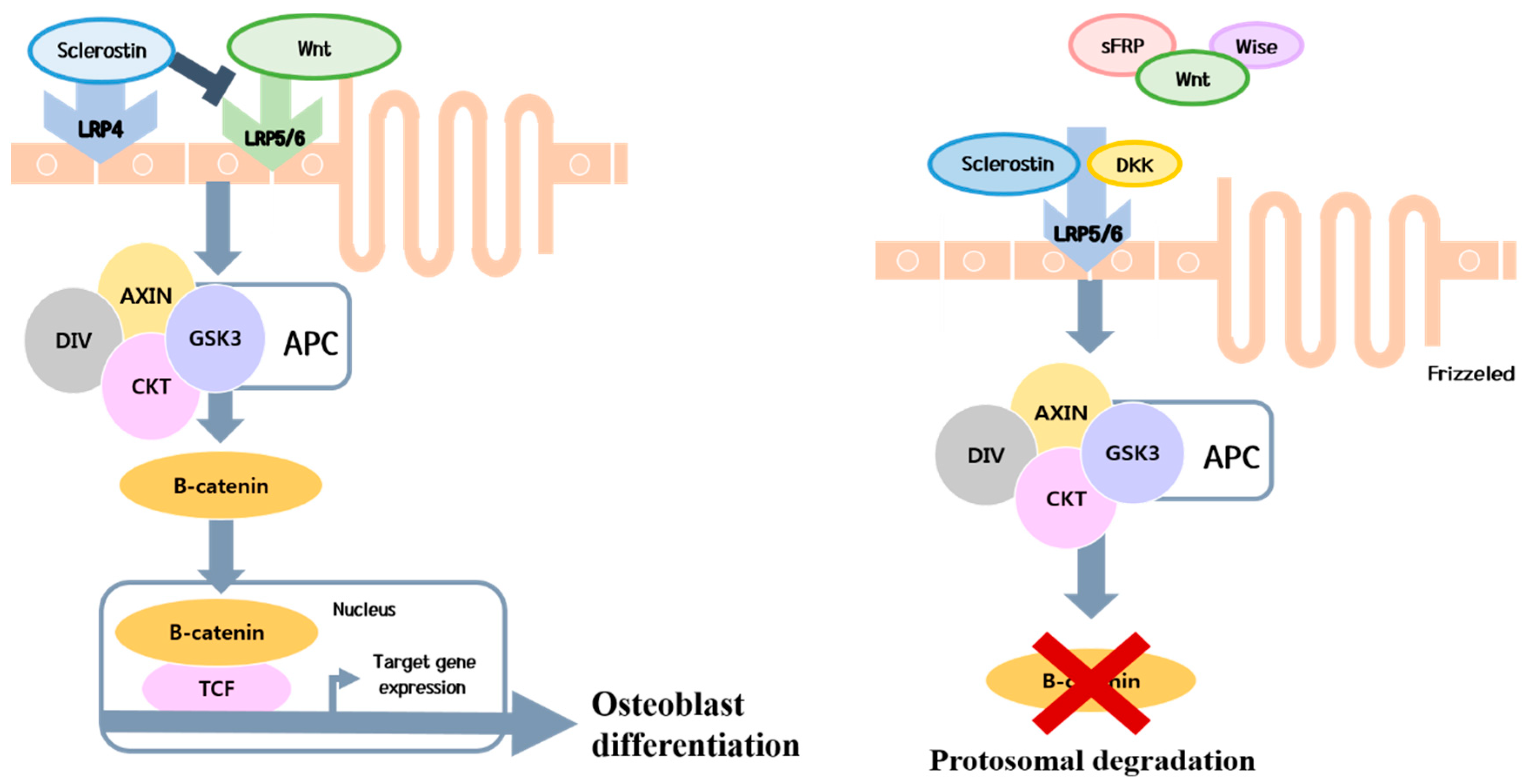
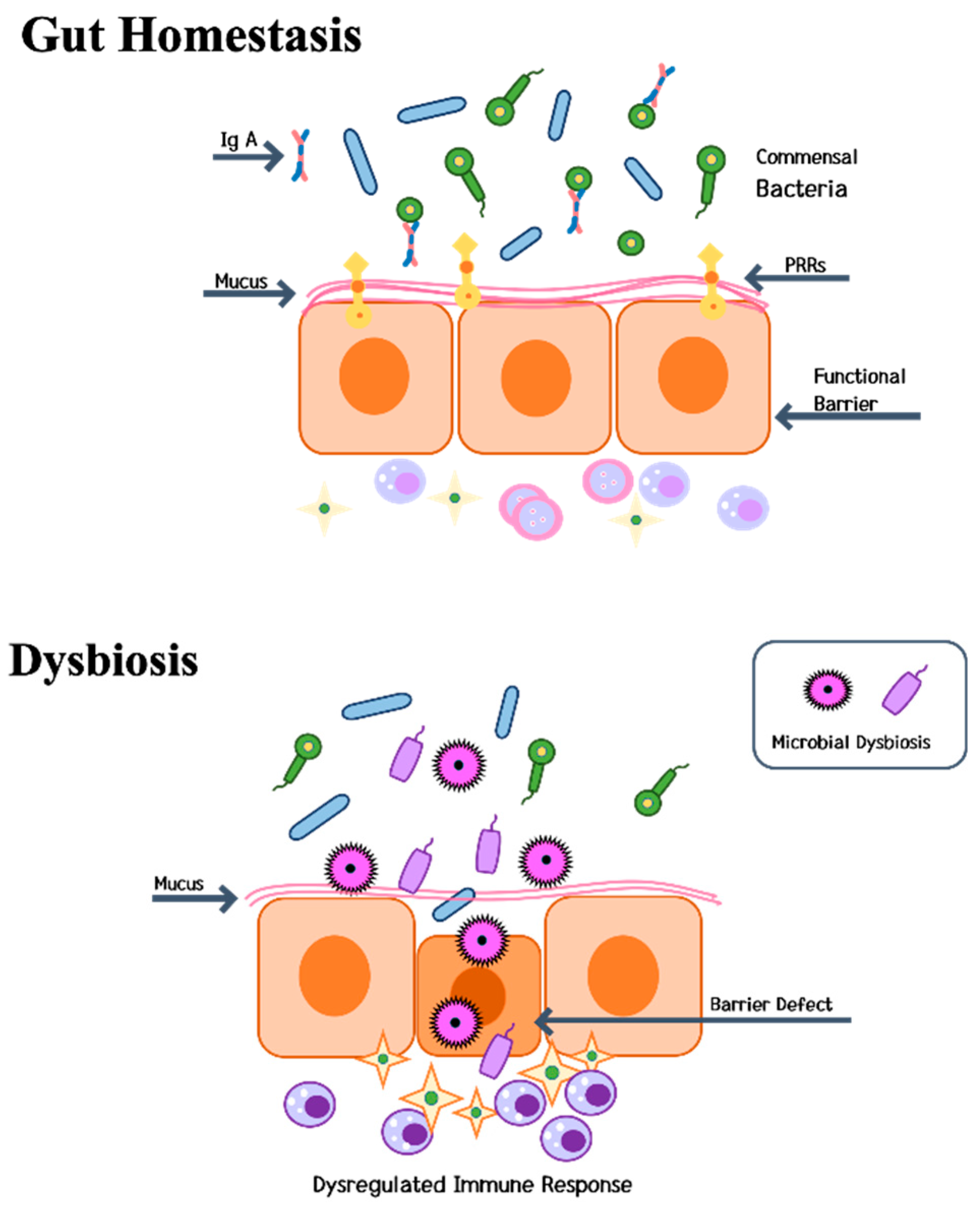
4. Pathogenesis
4.1. Predominant Decrease in Bone Formation
4.2. Predominant Increase in Bone Resorption
4.3. Gut-Liver-Bone Axis
5. Special Clinical Considerations
5.1. Bone Diseases in Patients with a Liver Transplant
5.2. Association between Chronic Hepatitis B Virus Infection Treatment and Bone Diseases
5.3. Association of Sarcopenia with Bone Diseases in Patients with Chronic Liver Disease
5.4. Association of Chronic Liver Disease with Avascular Necrosis of the Hip
6. Management
6.1. Approaches to Risk Management and Overall Nutritional Status
6.2. Pharmacological Therapies for Bone Diseases
6.3. Calcium and Vitamin D Supplementation
6.4. Pharmacological Therapies that Increase Bone Formation
6.5. Pharmacological Therapies that Decrease Bone Resorption
6.6. Therapies Targeting Gut Dysbiosis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nuti, R.; Brandi, M.L.; Checchia, G.; Di Munno, O.; Dominguez, L.; Falaschi, P.; Fiore, C.E.; Iolascon, G.; Maggi, S.; Michieli, R.; et al. Guidelines for the management of osteoporosis and fragility fractures. Intern. Emerg. Med. 2019, 14, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Burra, P.; Burroughsy, A.; Graziadei, I.; Pirenne, J.; Valdecasas, J.C.; Muiesan, P.; Samuel, D.; Forn, X. EASL Clinical Practice Guidelines: Liver transplantation. J. Hepatol. 2016, 64, 433–485. [Google Scholar]
- Compston, J.E.; McClung, M.R.; Leslie, W.D. Osteoporosis. Lancet 2019, 393, 364–376. [Google Scholar] [CrossRef]
- Collier, J. Bone disorders in chronic liver disease. Hepatology 2007, 46, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Guanabens, N.; Pares, A. Osteoporosis in chronic liver disease. Liver Int. 2018, 38, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Muñoz, S.J. Bone disease in cirrhosis. Clin. Liver Dis. 2015, 6, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Nakchbandi, I.A.; van der Merwe, S.W. Current understanding of osteoporosis associated with liver disease. Nat. Rev. Gastroenterol Hepatol. 2009, 6, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Pares, A.; Guanabens, N. Treatment of bone disorders in liver disease. J. Hepatol. 2006, 45, 445–453. [Google Scholar] [CrossRef]
- Collier, J.; Ninkovic, M.; Compston, J. Guidelines on the management of osteoporosis assciated with chronic liver disease. Gut 2002, 50, i1–i9. [Google Scholar] [CrossRef]
- Santos, L.A.; Romeiro, F.G. Diagnosis and Management of Cirrhosis-Related Osteoporosis. Biomed. Res. Int. 2016, 2016, 1423462. [Google Scholar] [CrossRef]
- Rouillard, S.; Lane, N.E. Hepatic osteodystrophy. Hepatology 2001, 33, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Guanabens, N.; Pares, A. Management of osteoporosis in liver disease. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 438–445. [Google Scholar] [CrossRef]
- Handzlik-Orlik, G.; Holecki, M.; Wilczyński, K.; Duława, J. Osteoporosis in liver disease: Pathogenesis and management. Ther. Adv. Endocrinol. Metab. 2016, 7, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.N.; Lie, J.D.; Wan, C.K.V.; Cameron, M.; Austel, A.G.; Nguyen, J.K.; Van, K.; Hyun, D. Osteoporosis: A Review of Treatment Options. P T. 2018, 43, 92–104. [Google Scholar] [PubMed]
- Eastell, R.; Rosen, C.J.; Black, D.M.; Cheung, A.M.; Murad, M.H.; Shoback, D. Pharmacological Management of Osteoporosis in Postmenopausal Women: An Endocrine Society* Clinical Practice Guideline. J. Clin. ENdocrinol. Metab. 2019, 104, 1595–1622. [Google Scholar] [CrossRef]
- Danford, C.J.; Trivedi, H.D.; Bonder, A. Bone Health in Patients With Liver Diseases. J. Clin. Densitom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Peck, W.A.; Burckhardt, P.; Christiansen, C.; Fleisch, H.A.; Genant, H.K.; Gennari, C.; Martin, T.J.; Martini, L.; Morita, R.; Ogata, E.; et al. Consensus development conference: Diagnosis, prophylaxis, and treatment of osteoporosis. Am. J. Med. 1993, 94, 646–650. [Google Scholar]
- Guanabens, N.; Pares, A. Liver and bone. Arch. Biochem. Biophys. 2010, 503, 84–94. [Google Scholar] [CrossRef]
- Camacho, P.M.; Petak, S.M.; Binkley, N.; Clarke, B.L.; Harris, S.T.; Hurley, D.L.; Kleerekoper, M.; Lewiecki, E.M.; Miller, P.D.; Narula, H.S.; et al. American Association of Clinical Endocrinologists and American College of Endocrinology Clinical Practice Guidelines for the Diagnosis and Treatment of Postmenopausal Osteoporosis. Endocr. Pract. 2016, 22, 1111–1118. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Beuers, U.; Corpechot, C.; Invernizzi, P.; Jones, D.; Marzioni, M.; Schramm, C. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Bering, T.; Diniz, K.G.D.; Coelho, M.P.P.; Vieira, D.A.; Soares, M.M.S.; Kakehasi, A.M.; Correia, M.I.T.D.; Teixeira, R.; Queiroz, D.M.M.; Rocha, G.A.; et al. Association between pre-sarcopenia, sarcopenia, and bone mineral density in patients with chronic hepatitis C. J. Cachexia Sarcopenia Muscle 2018, 9, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Carey, E.J.; Lai, J.C.; Wang, C.W.; Dasarathy, S.; Lobach, I.; Montano-Loza, A.J.; Dunn, M.A. A multicenter study to define sarcopenia in patients with end-stage liver disease. Liver Transpl. 2017, 23, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Moya-Angeler, J.; Gianakos, A.L.; Villa, J.C.; Ni, A.; Lane, J.M. Current concepts on osteonecrosis of the femoral head. World J. Orthop. 2015, 6, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Yatsonsky, l.D.; Pan, K.; Shendge, V.B.; Liu, J.; Ebraheim, N.A. Linkage of microbiota and osteoporosis: A mini literature review. World J. Orthop. 2019, 10, 123–127. [Google Scholar]
- Li, L.; Rao, S.; Cheng, Y.; Zhuo, X.; Deng, C.; Xu, N.; Zhang, H.; Yang, L. Microbial osteoporosis: The interplay between the gut microbiota and bones via host metabolism and immunity. MicrobiologyOpen. 2019, 8, e00810. [Google Scholar] [CrossRef] [PubMed]
- Schepper, J.D.; Collins, F.L.; Rios-Arce, N.D.; Raehtz, S.; Schaefer, L.; Gardinier, J.D.; Britton, R.A.; Parameswaran, N.; McCabe, L.R. Probiotic Lactobacillus reuteri Prevents Postantibiotic Bone Loss by Reducing Intestinal Dysbiosis and Preventing Barrier Disruption. J. Bone Miner. Res. 2019, 34, 681–698. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pederson, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- D’Amelio, P.; Sassi, F. Gut Microbiota, Immune System, and Bone. Calcif. Tissue Int. 2018, 102, 415–425. [Google Scholar] [CrossRef]
- Nakchbandi, I.A. Osteoporosis and fractures in liver disease: Relevance, pathogenesis and therapeutic implications. World J. Gastroenterol. 2014, 20, 9427–9438. [Google Scholar]
- Giouleme, O.I.; Vyzantiazdis, T.A.; Nikolaidis, N.L.; Vasiliadis, T.G.; Papageorgiou, A.A.; Eugenidis, N.P.; Harsoulis, F.I. Pathogenesis of Osteoporosis in Liver Cirrhosis. Hepato-Gastroenterology. 2006, 53, 938–943. [Google Scholar] [PubMed]
- Zheng, J.P.; Miao, H.X.; Zheng, S.W.; Liu, W.L.; Chen, C.Q.; Zhong, H.B.; Li, S.F.; Fang, T.P.; Sun, C.H. Risk factors for osteoporosis in liver cirrhosis patients measured by transient elastography. Medicine (Baltimore) 2018, 97, e10645. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.; Fevery, J.; Kalloo, A.; Nagorney, D.M.; Boberg, K.M.; Shneider, B.; Gores, J.G. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010, 51, 660–678. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.R.; Terrault, N.; Ojo, L.; Hay, J.E.; Neuberger, J.; Blumberg, E.; Teperman, L.W. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl. 2013, 19, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; DiMartini, A.; Feng, S.; Brown, R., Jr.; Fallon, M. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology 2014, 59, 1144–1165. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Chazouillères, O.; Dalekos, G.; Drenth, J.; Heneghan, M.; Hofer, H.; Lammert, F.; Lenzi, M. EASL Clinical Practice Guidelines: Autoimmune hepatitis. J. Hepatol. 2015, 63, 971–1004. [Google Scholar]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.; Chen, C.J.; Chen, D.S.; Chen, P.J.; Chien, R.N.; Dokmeci, A.K.; et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis; Zoulim, F. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019, 69, 394–419. [Google Scholar] [CrossRef] [PubMed]
- Merli, M.; Berzigotti, A.; Zelber-Sagi, S.; Dasarathy, S.; Montagnese, S.; Genton, L.; Plauth, M.; Pares, A. EASL Clinical Practice Guidelines on nutrition in chronic liver disease. J. Hepatol. 2019, 70, 172–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.M.; Lin, C.L.; Chang, S.N.; Sung, F.C.; Huang, S.T.; Kao, C.H. Osteoporosis and fractures after solid organ transplantation: A nationwide population-based cohort study. Mayo Clin Proc. 2014, 89, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Kaser, A.; Pines, A.; Dotan, I. Gut, inflammation and osteoporosis: Basic and clinical concepts. Gut 2008, 57, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Guss, J.D.; Taylor, E.; Rouse, Z.; Roubert, S.; Higgins, C.H.; Thomas, C.J.; Baker, S.P.; Vashishth, D.; Donelly, E.; Shea, M.K.; et al. The microbial metagenome and bone tissue composition in mice with microbiome-induced reductions in bone strength. Bone 2019, 127, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Schiefke, I.; Fach, A.; Wiedmann, M.; Aretin, A.V.; Schenker, E.; Borte, G.; Wiese, M.; Moessner, J. Reduced bone mineral density and altered bone turnover markers in patients with non-cirrhotic chronic hepatitis B or C infection. World J. Gastroenterol. 2005, 11, 1843–1847. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437–443. [Google Scholar] [CrossRef]
- Bonucci, E.; Ballanti, P. Osteoporosis-bone remodeling and animal models. Toxicol. Pathol. 2014, 42, 957–969. [Google Scholar] [CrossRef]
- Compston, J.E.; Vedi, S.; Kaptoge, S.; Seeman, E. Bone remodeling rate and remodeling balance are not co-regulated in adulthood: Implications for the use of activation frequency as an index of remodeling rate. J. Bone Miner. Res. 2007, 22, 1031–1036. [Google Scholar] [CrossRef]
- Wilson, T.; Katz, J.M.; Gray, D.H. Inhibition of active bone resorption by copper. Calcif Tissue Int. 1981, 33, 35–39. [Google Scholar] [CrossRef] [PubMed]
- De Vernejoul, M.C.; Pointillart, A.; Golenzer, C.C.; Morieux, C.; Bielakoff, J.; Modrowski, D.; Miravet, L. Effects of iron overload on bone remodeling in pigs. Am. J. Pathol. 1984, 116, 377–384. [Google Scholar] [CrossRef][Green Version]
- Diamond, T.; Pojer, R.; Stiel, D.; Alfrey, A.; Posen, S. Does iron affect osteoblast function? Studies in vitro and in patients with chronic liver disease. Calcif. Tissue Int. 1991, 48, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.L.; Bañon, S.; Andrés, R.; Perez-Elías, M.J.; Moreno, A.; Moreno, S. Prevalence of causes of secondary osteoporosis and contribution to lower bone mineral density in HIV infected patients. Osteoporos Int. 2014, 25, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Corazza, G.R.; Trevisani, F.; Di Stefano, M.; De Notariis, S.; Veneto, G.; Cecchetti, L.; Minguzzi, L.; Gasbarrini, G.; Bernardi, M. Early increase of bone resorption in patients with liver cirrhosis secondary to viral hepatitis. Dig. Dis. Sci. 2000, 45, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.P.; Kronenberger, B.; Bojunga, J.; Stamm, B.; Herrmann, E.; Bücker, A.; Mihm, U.; von Wagner, M.; Zeuzem, S.; Sarrazin, C. Prospective study of bone mineral density and metabolism in patients with chronic hepatitis C during pegylated interferon alpha and ribavirin therapy. J. Viral Hepat. 2008, 15, 790–796. [Google Scholar] [PubMed]
- Marie, P.J.; Kassem, M. Osteoblasts in osteoporosis: Past, emerging, and future anabolic targets. Eur. J. Endocrinol. 2011, 165, 1–10. [Google Scholar] [CrossRef]
- Ruiz-Gaspa, S.; Martinez-Ferrer, A.; Guanabens, N.; Dubreuil, M.; Peris, P.; Enjuanes, A.; Marinez de Osaba, M.J.; Alvarez, L.; Monegal, A.; Combalia, A.; et al. Effects of bilirubin and sera from jaundiced patients on osteoblasts: Contribution to the development of osteoporosis in liver diseases. Hepatology. 2011, 54, 2104–2113. [Google Scholar] [CrossRef]
- Valenti, L.; Varenna, M.; Fracanzani, A.L.; Rossi, V.; Fargion, S.; Sinigaglia, L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. 2009, 20, 549–555. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef]
- Reid, I.R. Targeting Sclerostin in Postmenopausal Osteoporosis: Focus on Romosozumab and Blosozumab. BioDrugs. 2017, 31, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.; Kim, W.J.; Han, K.J.; Lim, S.K.; Kim, S.H. Effect of liver dysfunction on circulating sclerostin. J. Bone Miner. Metab. 2014, 32, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Terasaka, T.; Iwata, N.; Katsuyama, T.; Komatsubara, M.; Nagao, R.; Inagaki, K.; Otsuka, F. Combined Effects of Androgen and Growth Hormone on Osteoblast Marker Expression in Mouse C2C12 and MC3T3-E1 Cells Induced by Bone Morphogenetic Protein. J. Clin. Med. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Crane, J.L.; Xie, L.; Xian, L.; Xie, H.; Cao, X. IGF-I induced phosphorylation of PTH receptor enhances osteoblast to osteocyte transition. Bone Res. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Menendez, L.; Sadaba, M.C.; Puche, J.E.; Lavandera, J.L.; de Castro, L.F.; de Gortazar, A.R.; Castilla-Cortázar, I. IGF-I increases markers of osteoblastic activity and reduces bone resorption via osteoprotegerin and RANK-ligand. J. Transl. Med. 2013, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, V.; Bianchi, V.E. Effect of GH/IGF-1 on Bone Metabolism and Osteoporsosis. Int. J. Endocrinol. 2014, 2014, 235060. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Invest. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Nasu, M.; Sugimoto, T.; Chihara, M.; Hiraumi, M.; Kurimoto, F.; Chihara, K. Effect of natural menopause on serum levels of IGF-I and IGF-binding proteins: Relationship with bone mineral density and lipid metabolism in perimenopausal women. Eu.r J. Endocrinol. 1997, 136, 608–616. [Google Scholar] [CrossRef]
- Cemborain, A.; Castilla-Cortazar, I.; Garcia, M.; Quiroga, J.; Muguerza, B.; Picardi, A.; Santidrián, S.; Prieto, J. Osteopenia in rats with liver cirrhosis: Beneficial effects of IGF-I treatment. J. Hepatol. 1998, 28, 122–131. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef]
- Liu, J.M.; Zhao, H.Y.; Ning, G.; Chen, Y.; Zhang, L.Z.; Sun, L.H.; Zhao, Y.J.; Xu, M.Y.; Chen, J.L. IGF-1 as an early marker for low bone mass or osteoporosis in premenopausal and postmenopausal women. J. Bone Miner. Metab. 2008, 26, 159–164. [Google Scholar] [CrossRef] [PubMed]
- De la Garza, R.G.; Morales-Garza, L.A.; Martin-Estal, I.; Castilla-Cortazar, I. Insulin-Like Growth Factor-1 Deficiency and Cirrhosis Establishment. J. Clin. Med. Res. 2017, 9, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Golds, G.; Houdek, D.; Arnason, T. Male Hypogonadism and Osteoporosis: The Effects, Clinical Consequences, and Treatment of Testosterone Deficiency in Bone Health. Int. J. Endocrinol. 2017, 2017, 4602129. [Google Scholar] [CrossRef] [PubMed]
- Seeman, E. The structural and biomechanical basis of the gain and loss of bone strength in women and men. Endocrinol. Metab. Clin. North. Am. 2003, 32, 25–38. [Google Scholar] [CrossRef]
- Moe, S.M. Calcium Homeostasis in Health and in Kidney Disease. Compr. Physiol. 2016, 6, 1781–1800. [Google Scholar] [PubMed]
- Cao, X. RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res. 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Nakchbandi, I.A.; Lang, R.; Kinder, B.; Insogna, K.L. The role of the receptor activator of nuclear factor-kappaB ligand/osteoprotegerin cytokine system in primary hyperparathyroidism. J. Clin. Endocrinol. Metab. 2008, 93, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Saini, V.; Pajevic, P.D. Effects of PTH on osteocyte function. Bone 2013, 54, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.; Kim, J.H.; Kim, N. Pro-inflammatory Cytokines Modulating Osteoclast Differentiation and Function. J. Rheum. Dis. 2016, 23, 148–153. [Google Scholar] [CrossRef]
- Guerrini, M.M.; Takayanagi, H. The immune system, bone and RANKL. Arch. Biochem. Biophys. 2014, 561, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Choi, Y. Biology of the RANKL–RANK–OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Cenci, S.; Weitzmann, M.N.; Roggia, C.; Namba, N.; Novack, D.; Woodring, J.; Pacifici, R. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J. Clin. Invest. 2000, 106, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.; Horowitz, M.; Choi, Y. Osteoimmunology: Interactions of the bone and immune system. Endocr. Rev. 2008, 29, 403–440. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.P.; Yao, Z.; Iida, M.H.; Clarke, C.M.; Doggett, B.; Xing, L.; Boyce, B.F. Malignant autosomal recessive osteopetrosis caused by spontaneous mutation of murine Rank. J. Bone Miner. Res. 2004, 19, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Titanji, K. Beyond Antibodies: B Cells and the OPG/RANK-RANKL Pathway in Health, Non-HIV Disease and HIV-Induced Bone Loss. Front. Immunol. 2017, 8, 1851. [Google Scholar] [CrossRef]
- Naseem, S.; Hussain, T.; Manzoor, S. Interleukin-6: A promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev. 2018, 39, 36–45. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Nakchbandi, I.A.; Mitnick, M.A.; Lang, R.; Gundberg, C.; Kinder, B.; Insogna, K. Circulating levels of interleukin-6 soluble receptor predict rates of bone loss in patients with primary hyperparathyroidism. J. Clin. Endocrinol. Metab. 2002, 87, 4946–4951. [Google Scholar] [CrossRef][Green Version]
- Blaschke, M.; Koepp, R.; Cortis, J.; Komrakova, M.; Schieker, M.; Hempel, U.; Siggelkow, H. IL-6, IL-1beta, and TNF-alpha only in combination influence the osteoporotic phenotype in Crohn’s patients via bone formation and bone resorption. Adv. Clin. Exp. Med. 2018, 27, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Z.Y.; Zhang, Y.Y.; Yang, H.L. IL-6 Contributes to the Defective Osteogenesis of Bone Marrow Stromal Cells from the Vertebral Body of the Glucocorticoid-Induced Osteoporotic Mouse. PLoS ONE 2016, 11, e0154677. [Google Scholar] [CrossRef] [PubMed]
- Axmann, R.; Bohm, C.; Kronke, G.; Zwerina, J.; Smolen, J.; Schett, G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009, 60, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.A.; He, M.; Kang, L.I.; Ding, M.Q.; Radder, J.E.; Haynes, M.M.; Yang, Y.; Paranjpe, S.; Bowen, W.C.; Orr, A.; et al. Synthesis of IL-6 by Hepatocytes Is a Normal Response to Common Hepatic Stimuli. PLoS ONE 2014, 9, e96053. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Matsumata, T.; Taketomi, A.; Shirabe, K.; Yamamoto, K.; Takenaka, K.; Sugimachi, K. The role of interleukin-6, interleukin-16, tumor necrosis factor-alpha and endotoxin in hepatic resection. Hepatogastroenterology 1995, 42, 691–697. [Google Scholar] [PubMed]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Eskan, M.A.; Benakanakere, M.R.; Rose, B.G.; Zhang, P.; Zhao, J.; Stathopoulou, P.; Fujioka, D.; Kinane, D.F. Interleukin-1beta modulates proinflammatory cytokine production in human epithelial cells. Infect. Immun. 2008, 76, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Cipriani, P.; Carubbi, F.; Liakouli, V.; Zazzeroni, F.; Di Benedetto, P.; Berardicurti, O.; Alesse, E.; Giacomelli, R. The role of IL-1beta in the bone loss during rheumatic diseases. Mediators Inflamm. 2015, 2015, 782382. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Nakamura, I.; Jimi, E. Regulation of osteoclast differentiation and function by interleukin-1. Vitam. Horm. 2006, 74, 357–370. [Google Scholar]
- Lee, Y.M.; Fujikado, N.; Manaka, H.; Yasuda, H.; Iwakura, Y. IL-1 plays an important role in the bone metabolism under physiological conditions. Int. Immunol. 2010, 22, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Grimes, S.N.; Li, S.; Hu, X.; Ivashkiv, L.B. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J Exp. Med. 2012, 209, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Li, P.; Yao, Z.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; O’Keefe, R.J.; Xing, L. TNF-alpha and pathologic bone resorption. Keio J. Med. 2005, 54, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Yuen, T.; Sun, L.; Zaidi, M. From the gut to the strut: Where inflammation reigns, bone abstains. J. Clin Invest. 2016, 126, 2045–2048. [Google Scholar] [CrossRef]
- Pazianas, M.; Rhim, A.D.; Weinberg, A.M.; Su, C.; Lichtenstein, G.R. The effect of anti-TNF-alpha therapy on spinal bone mineral density in patients with Crohn’s disease. Ann. N. Y. Acad. Sci. 2006, 1068, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Lai, J.C.; Sawinski, D.; Zeigler, T.E.; Cedars, M.; Forde, K.A. Sex hormone levels by presence and severity of cirrhosis in women with chronic hepatitis C virus infection. J. Viral Hepat. 2019, 26, 258–262. [Google Scholar] [CrossRef]
- Zumoff, B.; Fishman, J.; Gallagher, T.F.; Hellman, L. Estradiol metabolism in cirrhosis. J. Clin. Invest. 1968, 47, 20–25. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vierira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, C.P.; Neut, C.; Desreumaux, P.; Colombel, J.F. Dysbiosis in inflammatory bowel disease. Gut. 2004, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Greenbaum, J.; Shen, H.; Deng, H.W. Association between gut microbiota and bone health: Potential mechanisms and prospective. J. Clin. Endocrinol. Metab. 2017, 102, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Young, V.B. The role of the microbiome in human health and disease: An introduction for clinicians. BMJ 2017, 356, j831. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Pacifici, R.; Rifas, L.; Teitelbaum, S.; Slatopolsky, E.; McCracken, R.; Bergfeld, M.; Lee, W.; Avioli, L.V.; Peck, W.A. Spontaneous release of interleukin 1 from human blood monocytes reflects bone formation in idiopathic osteoporosis. Proc. Natl. Acad. Sci. 1987, 84, 4616–4620. [Google Scholar] [CrossRef]
- Sjogren, K.; Engdahl, C.; Henning, P.; Lerner, U.H.; Tremaroli, V.; Lagerquist, M.K.; Bäckhed, F.; Ohlsson, C. The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 2012, 27, 1357–1367. [Google Scholar] [CrossRef]
- Novince, C.M.; Whittow, C.R.; Aartun, J.D.; Hathaway, J.D.; Poulides, N.; Chavez, M.B.; Steinkamp, H.M.; Kirkwood, K.A.; Huang, E.; Westwater, C. Commensal gut microbiota immunomodulatory actions in bone marrow and liver have catabolic effects on skeletal homeostasis in health. Sci. Rep. 2017, 7, 5747. [Google Scholar] [CrossRef]
- Ohlsson, C.; Sjogren, K. Effects of the gut microbiota on bone mass. Trends Endocrinol. Metab. 2015, 26, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Pacifici, R. From osteoimmunology to osteomicrobiology: How the microbiota and the immune system regulate bone. Calcif. Tissue Int. 2018, 102, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Chassaing, B.; Tyagi, A.M.; Vaccaro, C.; Luo, T.; Adams, J.; Darby, T.M.; Weitzmann, M.N.; Mulle, J.G.; Gewirtz, A.T.; et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Invest. 2016, 126, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Blanton, L.V.; Charbonneau, M.R.; Salih, T.; Barratt, M.J.; Venkatesh, S.; Ilkaveya, O.; Subramanian, S.; Manary, M.J.; Trehan, I.; Jorgensen, J.M.; et al. Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science. 2016, 351, aad3311. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, L.; Rouleau, M.; Wakkach, A.; Blin-Wakkach, C. Gut microbiome and bone. Jt. Bone Spine. 2019, 86, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, M.; Makki, K.; Storelli, G.; Machuca-Gayet, I.; Srutkova, D.; Hermanova, P.; Martino, M.E.; Balmand, S.; Hudcovic, T.; Heddi, A.; et al. Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition. Science 2016, 351, 854–857. [Google Scholar] [CrossRef]
- Yan, J.; Charles, J.F. Gut microbiome and bone: To build, destroy, or both? Curr. Osteoporos. Rep. 2017, 15, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Herzog, J.W.; Tsang, K.; Brennan, C.A.; Bower, M.A.; Garrett, W.S.; Sartor, B.R.; Aliprantis, A.O.; Charles, J.F. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 2016, 113, E7554–E7563. [Google Scholar] [CrossRef]
- Das, M.; Cronin, O.; Keohane, D.M.; Cormac, E.M.; Nugent, H.; Nugent, M.; Molloy, C.; O’Toole, P.W.; Shanahan, F.; Molloy, M.G.; et al. Gut microbiota alterations associated with reduced bone mineral density in older adults. Rheumatology 2019. [Google Scholar] [CrossRef]
- Pacifici, R. Bone Remodeling and the Microbiome. Cold Spring Harb Perspect Med. 2018, 8, a031203. [Google Scholar] [CrossRef]
- Nilsson, A.G.; Sundh, D.; Backhed, F.; Lorentzon, M. Lactobacillus reuteri reduces bone loss in older women with low bone mineral density: A randomized, placebo-controlled, double-blind, clinical trial. J. Intern. Med. 2018, 284, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Whisner, C.M.; Castillo, L.F. Prebiotics, bone and mineral metabolism. Calcif. Tissue Int. 2018, 102, 443–479. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R. Nutritional influence on bone: Role of gut microbiota. Aging Clin. Exp. Res. 2019, 31, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Chascsa, D.M.; Vargas, H.E. The gastroenterologist’s guide to management of the post-liver transplant patient. Am. J. Gastroenterol. 2018, 113, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.B.; Xie, X.B.; Peng, L.K.; Liu, L.; Song, L.; Dai, H.L. Current status of research on osteoporosis after solid organ transplantation: Pathogenesis and management. Biomed. Res. Int. 2015, 2015, 413169. [Google Scholar] [CrossRef] [PubMed]
- Anastasilakis, A.D.; Tsourdi, E.; Makras, P.; Polyzos, S.A.; Meier, C.; McCloskey, E.V.; Pepe, J.; Zillikens, M.C. Bone disease following solid organ transplantation: A narrative review and recommendations for management from The European Calcified Tissue Society. Bone 2019, 127, 401–418. [Google Scholar] [CrossRef]
- Elizabeth, B.H.; Cornelis, J.P.T.; Post, J.G.; Slooff, M.J.H.; Gips, C.H. Bone disease after orthotopic liver transplantation. J. Hepatol. 1988, 6, 94–100. [Google Scholar]
- Ninkovic, M.; Love, S.A.; Tom, B.; Alexander, G.J.; Compston, J.E. High prevalence of osteoporosis in patients with chronic liver disease prior to liver transplantation. Calcif. Tissue Int. 2001, 69, 321–326. [Google Scholar] [CrossRef]
- Loria, I.; Albanese, C.; Giusto, M.; Galtieri, P.A.; Giannelli, V.; Lucidi, C.; Di Menna, S.; Pirazzi, C.; Corradini, S.G.; Mennini, G. Bone disorders in patients with chronic liver disease awaiting liver transplantation. Transplant. Proc. 2010, 42, 1191–1193. [Google Scholar] [CrossRef]
- Guichelaar, M.M.; Malinchoc, M.; Sibonga, J.D.; Clarke, B.L.; Hay, J.E. Bone histomorphometric changes after liver transplantation for chronic cholestatic liver disease. J. Bone Miner. Res. 2003, 18, 2190–2199. [Google Scholar] [CrossRef]
- Hay, J.E. Osteoporosis in liver diseases and after liver transplantation. J. Hepatol. 2003, 38, 856–865. [Google Scholar] [CrossRef]
- Verdi, S.; Greer, S.; Skingle, S.g.; Garrahan, N.J.; Ninkovic, M.; Alexander, G.A.; Compston, J.E. Mechanism of bone loss after liver transplantation: A histomorphometric analysis. J. Bone Miner. Res. 1999, 14, 281–287. [Google Scholar]
- Song, L.; Xie, X.B.; Peng, L.K.; Yu, S.J.; Peng, Y.T. Mechanism and treatment strategy of osteoporosis after transplantation. Int. J. Endocrinol. 2015, 2015, 280164. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Hawkins, M.; Steinberg, K.; Hersh, T.; Galambos, J.T.; Henderson, J.M.; Millikan, W.J.; Galloway, J.R. Prevalence and prediction of osteopenia in chronic liver disease. Hepatology 1990, 12, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.H.; Oldroyd, B.; Stewart, S.P.; Roman, F.; Smith, M.A.; Pollard, S.; Lodge, P.; O’Grady, J.G.; Losowsky, M.S. Regional bone mineral density after orthotopic liver transplantation. Eur. J. Gastroenterol. Hepatol. 1999, 11, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, P.W.; Bischoff-Ferrari, H.A.; Boggian, K.; Bonani, M.; van Delden, C.; Enriquez, N.; Fehr, T.; Garzoni, C.; Hirsch, H.H.; Hirzel, C.; et al. Bone metabolism dynamics in the early post-transplant period following kidney and liver transplantation. PLoS ONE 2018, 13, e0191167. [Google Scholar] [CrossRef]
- Monegal, A.; Navasa, M.; Guanabens, N.; Peris, P.; Pons, F.; Martinez de Osaba, M.J.; Ordi, J.; Rimola, A.; Rodés, J.; Muñoz-Gómez, J. Bone disease after liver transplantation: A long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos. Int. 2001, 12, 484–492. [Google Scholar] [CrossRef]
- Mukherjee, S.; Mukherjee, U. A comprehensive review of immunosuppression used for liver transplantation. J. Transplant. 2009, 2009, 701464. [Google Scholar] [CrossRef]
- Compston, J.; Shane, E. Bone Disease of Organ. Transplantation, 1st ed.; Elsevier Academic Press: Cambridge, MA, USA, 2005; pp. 271–285. [Google Scholar]
- Kulak, C.A.; Borba, V.Z.; Kulak, J., Jr.; Custodio, M.R. Osteoporosis after transplantation. Curr. Osteoporos. Rep. 2012, 10, 48–55. [Google Scholar] [CrossRef]
- Briot, K.; Roux, C. Glucocorticoid-induced osteoporosis. RMD Open. 2015, 1, e000014. [Google Scholar] [CrossRef]
- Buckley, L.; Humphrey, M.B. Glucocorticoid-Induced Osteoporosis. N. Engl. J. Med. 2018, 379, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, I.F.; Pham, L.; Mansky, L.M.; Gopalakrishnan, R.; Kim, C.M. Tenofovir-associated bone density loss. Ther. Clin. Risk Manag. 2010, 6, 41–47. [Google Scholar]
- Casado, J.L.; Santiuste, C.; Vazquez, M.; Banon, S.; Rosillo, M.; Gomez, A.; Gomez, A.; Perez-Elías, M.J.; Caballero, C.; Rey, J.M.; et al. Bone mineral density decline according to renal tubular dysfunction and phosphaturia in tenofovir-exposed HIV-infected patients. AIDS 2016, 30, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, J.I.; Mocroft, A.; Mallon, P.W.; Wallet, C.; Gerstoft, J.; Russell, C.; Reiss, P.; Katlama, C.; De Wit, S.; Richert, L.; et al. Bone mineral density and inflammatory and bone biomarkers after darunavir–ritonavir combined with either raltegravir or tenofovir–emtricitabine in antiretroviral-naive adults with HIV-1: A substudy of the NEAT001/ANRS143 randomised trial. Lancet HIV. 2015, 2, e464–e473. [Google Scholar] [CrossRef]
- Komatsu, A.; Ikeda, A.; Kikuchi, A.; Minami, C.; Tan, M.; Matsushita, S. Osteoporosis-related fractures in HIV-infected patients receiving long-term tenofovir disoproxil fumarate: An observational cohort study. Drug Saf. 2018, 41, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.T.; Le, A.K.; Chang, M.S.; Hsu, H.; Nguyen, P.; Zhang, J.Q.; Wong, C.; Cheung, R.; Nguyen, M.H. Antiviral therapy and the development of osteopenia/osteoporosis among Asians with chronic hepatitis B. J. Med. Virol. 2019, 91, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.M.; Cotter, A.G. Tenofovir and bone health. Curr. Opin. HIV AIDS. 2016, 11, 326–332. [Google Scholar] [CrossRef]
- Gallant, J.E.; Staszewski, S.; Pozniak, A.L.; DeJesus, E.; Suleiman, J.M.A.H.; Miller, M.D.; Coakley, D.F.; Lu, B.; Toole, J.J.; Cheng, A.K.; et al. Efficacy and safety of tenofovir DF vs. stavudine in combination therapy in antiretroviral-naïve patients a 3-year randomized trial. JAMA 2004, 292, 191–201. [Google Scholar] [CrossRef]
- Fontana, R.J. Side effects of long-term oral antiviral therapy for hepatitis B. Hepatology 2009, 49, 185–195. [Google Scholar] [CrossRef]
- Gill, U.S.; Zissimopoulos, A.; Al-Shamma, S.; Burke, K.; McPhail, M.J.; Barr, D.A.; Kallis, Y.N.; Marley, R.T.; Foster, G.R.; Kenedu, P.T. Assessment of bone mineral density in tenofovir-treated patients with chronic hepatitis B: Can the fracture risk assessment tool identify those at greatest risk? J. Infect. Dis. 2015, 211, 374–382. [Google Scholar] [CrossRef]
- Bedimo, R.; Maalouf, N.M.; Zhang, S.; Drechsler, H.; Tebas, P. Osteoporotic fracture risk associated with cumulative exposure to tenofovir and other antiretroviral agents. AIDS 2012, 26, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.; Kwan, P.; Fabri, M.; Horban, A.; Pelemis, M.; Hann, H.W.; Gruel, S.; Caruntu, F.A.; Flaherty, G.F.; Massetto, B.; et al. Randomized comparison of tenofovir disoproxil fumarate vs. emtricitabine and tenofovir disoproxil fumarate in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology 2014, 146, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Cote, H.C. Mechanisms of antiretroviral therapy-induced mitochondrial dysfunction. Curr. Opin. HIV AIDS. 2007, 2, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Fux, C.A.; Rauch, A.; Simcock, M.; Bucher, H.C.; Hirschel, B.; Opravil, M.; Vernazz, P.; Cavassini, M.; Vernasconi, E.; Elzi, L.; et al. Tenofovir use is associated with an increase in serum alkaline phosphatase in the Swiss HIV Cohort Study. Antivir. Ther. 2008, 13, 1077–1082. [Google Scholar] [PubMed]
- Kohler, J.J.; Hosseini, S.H.; Hoying-Brandt, A.; Green, E.; Johnson, D.M.; Russ, R.; Tran, D.; Raper, C.M.; Santoianni, R.; Lewis, M. Tenofovir renal toxicity targets mitochondria of renal proximal tubules. Lab. Invest. 2009, 89, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Mothobi, N.Z.; Masters, J.; Marriott, D.J. Fanconi syndrome due to tenofovir disoproxil fumarate reversed by switching to tenofovir alafenamide fumarate in an HIV-infected patient. Ther. Adv. Infect. Dis. 2018, 5, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Locquet, M.; Beaudart, C.; Bruyere, O.; Kanis, J.A.; Delandsheere, L.; Reginster, J.Y. Bone health assessment in older people with or without muscle health impairment. Osteoporos. Int. 2018, 29, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J.; Mullen, K.D.; Kalhan, S.C. Measurements of total body and extracellular water in cirrhotic patients with and without ascites. Hepatology. 1991, 14, 1102–1111. [Google Scholar] [CrossRef]
- Owen, O.E.; Reichle, F.A.; Mozzoli, M.A.; Kreulen, T.; Patel, M.S.; Elfenbein, I.B.; Golsorkhi, M.; Chang, K.H.; Rao, N.S.; Sue, H.S.; et al. Hepatic, gut, and renal substrate flux rates in patients with hepatic cirrhosis. J. Clin. Invest. 1981, 68, 240–252. [Google Scholar] [CrossRef]
- Bansal, R.K.; Kumar, M.; Sachdeva, P.R.M.; Kumar, A. Prospective study of profile of hepatic osteodystrophy in patients with non-choleastatic liver cirrhosis and impact of bisphosphonate supplementation. United Eur. Gastroenterol. J. 2016, 4, 77–83. [Google Scholar] [CrossRef]
- Ormsbee, M.J.; Prado, C.M.; Ilich, J.Z.; Purcell, S.; Siervo, M.; Folsom, A.; Panton, L. Osteosarcopenic obesity: The role of bone, muscle, and fat on health. J. Cachexia Sarcopenia Muscle. 2014, 5, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Ilich, J.Z.; Brownbill, R.A. Habitual and low-impact activities are associated with better bone outcomes and lower body fat in older women. Calcif. Tissue Int. 2008, 83, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Ilich-Ernst, J.; Brownbill, R.A.; Ludemann, M.A.; Fu, R. Critical factors for bone health in women across the age span: How important is muscle mass? Medscape Women’s Health 2002, 7, 2. [Google Scholar] [PubMed]
- Patel, H.P.; Dawson, A.; Westbury, L.D.; Hasnaoui, G.; Syddall, H.E.; Shaw, S.; Sayer, A.A.; Cooper, C.; Dennison, E.M. Muscle mass, muscle morphology and bone health among community-dwelling older men: Findings from the Hertfordshire Sarcopenia Study (HSS). Calcif. Tissue Int. 2018, 103, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Buckinx, F.; Landi, F.; Cesari, M.; Fielding, R.A.; Visser, M.; Engelke, K.; Maggi, S.; Dennison, E.; Al-Daghri, N.M.; Allepaerts, S.; et al. Pitfalls in the measurement of muscle mass: A need for a reference standard. J. Cachexia Sarcopenia Muscle. 2018, 9, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.E.; Williams, D.; Lee, M.J. Osteoporosis, obesity, and sarcopenia on abdominal CT: A review of epidemiology, diagnostic criteria, and management strategies for the reporting radiologist. Abdom. Radiol. 2017, 42, 2376–2386. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Moncada, S. Hyperdynamic circulation in cirrhosis: A role for nitric oxide? Lancet. 1991, 337, 776–778. [Google Scholar] [CrossRef]
- Deleuran, T.; Overgaard, S.; Vilstrup, H.; Jepsen, P. Cirrhosis is a risk factor for total hip arthroplasty for avascular necrosis. Acta. Orthop. 2016, 87, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tsai, I.F.; Ho, Y.P.; Huang, C.T.; Lin, Y.C.; Lin, C.J.; Tseng, S.C.; Lin, W.P.; Chen, W.T.; Sheen, I.S. Endotoxemia contributes to the immune paralysis in patients with cirrhosis. J. Hepatol. 2007, 46, 816–826. [Google Scholar] [CrossRef]
- Albillos, A.; Lario, M.; Alvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Northup, P.G.; Caldwell, S.H. Coagulation in liver disease: A guide for the clinician. Clin. Gastroenterol. Hepatol. 2013, 11, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.L.; Caldwell, S.H. Assessing the Risk of Bleeding and Clotting in Cirrhosis. Clin. Liver Dis. 2016, 7, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Hsieh, Y.H.; Tsai, C.C.; Tseng, C.W.; Tseng, K.C.; Tsai, C.C. Is liver cirrhosis a risk factor for osteonecrosis of the femoral head in adults? A population-based 3-year follow-up study. Intern. Med. 2011, 50, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Morse, C.G.; Dodd, L.E.; Nghiem, K.; Costello, R.; Csako, G.; Lane, H.C.; Lozier, J.N.; Kovacs, J.A. Elevations in D-dimer and C-reactive protein are associated with the development of osteonecrosis of the hip in HIV-infected adults. AIDS 2013, 27, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Marvie, P.; Lisbonne, M.; L’Helgoualc’h, A.; Rauch, M.; Turlin, B.; Preisser, L.; Bourd-Boittin, K.; Théret, N.; Gascan, H.; Piquet-Pellorce, C.; et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J. Cell Mol. Med. 2010, 14, 1726–1739. [Google Scholar] [CrossRef] [PubMed]
- Bémeur, C.; Butterworth, R.F. Nutrition in the management of cirrhosis and its neurological complications. J. Clin. Exp. Hepatol. 2014, 4, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Marzocchi, R.; Agostini, F.; Marchesini, G. Update on branched-chain amino acid supplementation in liver diseases. Curr. Opin. Gastroenterol. 2005, 21, 197–200. [Google Scholar] [CrossRef]
- Shergill, R.; Syed, W.; Rizvi, S.A.; Singh, I. Nutritional support in chronic liver disease and cirrhotics. World J. Hepatol. 2018, 10, 685–694. [Google Scholar] [CrossRef]
- Shiomi, S.; Masaki, K.; Habu, D.; Takeda, T.; Nishiguchi, S.; Kuroki, T.; Tanaka, T.; Ochi, H. Calcitriol for bone disease in patients with cirrhosis of the liver. J. Gastroenterol. Hepatol. 1999, 14, 547–552. [Google Scholar] [CrossRef]
- Castelo-Branco, C.; Cortes, X.; Ferrer, M. Treatment persistence and compliance with a combination of calcium and vitamin D. Climacteric. 2010, 13, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Bolland, M.J.; Leung, W.; Tai, V.; Bastin, S.; Gamble, G.D.; Grey, A.; Reid, I.R. Calcium intake and risk of fracture: Systematic review. BMJ. 2015, 351, h4580. [Google Scholar] [CrossRef] [PubMed]
- Kleerekoper, M.; Mendlovic, D.B. Sodium fluoride therapy of postmenopausal osteoporosis. Endocr. Rev. 1993, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Guanabens, N.; Pares, A.; del Rio, L.; Roca, M.; Gomez, R.; Munoz, J.; Rodes, J. Sodium fluoride prevents bone loss in primary biliary cirrhosis. J. Hepatol. 1992, 15, 345–349. [Google Scholar] [CrossRef]
- Canalis, E. Update in new anabolic therapies for osteoporosis. J. Clin. Endocrinol. Metab. 2010, 95, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.R.; Ko, N.Y.; Chen, K.H. Medical treatment for osteoporosis: From molecular to clinical opinions. Int. J. Mol. Sci. 2019, 20, 2213. [Google Scholar] [CrossRef]
- Yurci, A.; Kalkan, A.O.; Ozbakir, O.; Karaman, A.; Torun, E.; Kula, M.; Baskol, M.; Gursoy, S.; Yucesoy, M.; Bayram, F. Efficacy of different therapeutic regimens on hepatic osteodystrophy in chronic viral liver disease. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1206–1212. [Google Scholar] [CrossRef]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef]
- Sieber, P.; Lardelli, P.; Kraenzlin, C.A.; Kraenzlin, M.E.; Meier, C. Intravenous bisphosphonates for postmenopausal osteoporosis: Safety profiles of zoledronic acid and ibandronate in clinical practice. Clin. Drug Investig. 2013, 33, 117–122. [Google Scholar] [CrossRef]
- Atamaz, F.; Hepguler, S.; Akyildiz, M.; Karasu, Z.; Kilic, M. Effects of alendronate on bone mineral density and bone metabolic markers in patients with liver transplantation. Osteoporos. Int. 2006, 17, 942–949. [Google Scholar] [CrossRef]
- Dodidou, P.; Bruckner, T.; Hosch, S.; Haass, M.; Klar, E.; Sauer, P.; Ziegler, R.; Leidig-Bruckner, G. Better late than never? Experience with intravenous pamidronate treatment in patients with low bone mass or fractures following cardiac or liver transplantation. Osteoporos. Int. 2003, 14, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Safer, U.; Safer, V.B.; Demir, S.O.; Yanikoglu, I. Effects of bisphosphonates and calcium plus vitamin-D supplements on cognitive function in postmenopausal osteoporosis section sign. Endocr. Metab. Immune Disord. Drug Targets. 2016, 16, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.M.; Bodingbauer, M.; Roschger, P.; Wekerle, T.; Pakrah, B.; Haas, M.; Kainz, A.; Oberbauer, R.; Muhlbacher, F.; Klasushoger, K. Short-term effects of high-dose zoledronic acid treatment on bone mineralization density distribution after orthotopic liver transplantation. Calcif. Tissue Int. 2008, 83, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Danford, C.J.; Trivedi, H.D.; Papamichael, K.; Tapper, E.B.; Bonder, A. Osteoporosis in primary biliary cholangitis. World J. Gastroenterol. 2018, 24, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Bilezikian, J.P.; Dempster, D.W.; Lewiecki, E.M.; Miller, P.D.; Neer, R.M.; Recker, R.R.; Shane, E.; Shoback, D.; Potts, J.T. Benefits and risks of bisphosphonate therapy for osteoporosis. J. Clin. Endocrinol. Metab. 2012, 97, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Conte, P.; Guarneri, V. Safety of intravenous and oral bisphosphonates and compliance with dosing regimens. Oncologist. 2004, 9, 28–37. [Google Scholar] [CrossRef] [PubMed]
- An, K.C. Selective Estrogen receptor modulators. Asian Spine J. 2016, 10, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Harnois, D.M.; Angulo, P.; Jorgensen, R.; Lindor, K.D. Raloxifene improves bone mass in osteopenic women with primary biliary cirrhosis: Results of a pilot study. Liver Int. 2005, 25, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Um, M.J.; Cho, E.A.; Jung, H. Combination therapy of raloxifene and alendronate for treatment of osteoporosis in elderly women. J. Menopausal Med. 2017, 23, 56–62. [Google Scholar] [CrossRef]
- Gambacciani, M.; Levancini, M. Hormone replacement therapy and the prevention of postmenopausal osteoporosis. Prz Menopauzalny. 2014, 13, 213–220. [Google Scholar]
- Sugimoto, T. Anti-RANKL monoclonal antibody denosumab (AMG162). Clin. Calcium. 2011, 21, 46–51. [Google Scholar] [PubMed]
- Malnick, S.; Maor, Y.; Melzer, E.; Ziv-Sokolowskaia, N.N.; Neuman, M.G. Severe hepatocytotoxicity linked to denosumab. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 78–85. [Google Scholar] [PubMed]
- Hernandez, C.J.; Guss, J.D.; Luna, M.; Goldring, S.R. Links between the microbiome and bone. J. Bone Miner. Res. 2016, 31, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Naik, S. Compartmentalized and systemic control of tissue immunity by commensals. Nat. Immunol. 2013, 14, 646–653. [Google Scholar] [CrossRef]
- Mbalaviele, G.; Novack, D.V.; Schett, G.; Teitelbaum, S.L. Inflammatory osteolysis: A conspiracy against bone. J. Clin. Invest. 2017, 127, 2030–2039. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Guidelines or Guidance | Bone Diseases Related to Liver Disease | |
|---|---|---|
| EASL 1 | 2018 Clinical Practice Guidelines: Nutrition in chronic liver disease [41] | Nutritional treatment options in cirrhotic patients with bone diseases |
| EASL 1 | 2017 Clinical Practice Guidelines: Management of hepatitis B virus infection [38] | Indications for selecting entecavir or tenofovir alafenamide over tenofovir disoproxil fumarate |
| EASL 1 | 2017 Clinical Practice Guidelines: Primary biliary cholangitis [20] | Management of complications: osteoporosis |
| EASL 1 | 2016 Clinical Practice Guidelines: Liver transplantation [2] | Bone disease screening and management |
| EASL 1 | 2015 Clinical Practice Guidelines: Autoimmune hepatitis [36] | Osteopenia/osteoporosis screening and management |
| AASLD 2 | 2018 Primary Biliary Cholangitis: Practice guidance [40] | Complication related to chronic cholestasis: osteoporosis/osteopenia management |
| AASLD 2 | 2018 Hepatitis B Guidance: Update on prevention, diagnosis and treatment of chronic hepatitis B [39] | Tenofovir disoproxil fumarate-associated bone disease |
| AASLD 2 | 2013 Evaluation for Liver Transplantation in Adults: Practice guideline [35] | Bone densitometry as part of transplant evaluation and treatment of osteoporosis initiated prior to liver transplantation |
| AASLD 2 | 2012 Practice guidelines by AASLD and AST 3: Long-term management of the successful adult liver transplant [34] | Bone mineral density follow up and management |
| AASLD 2 | 2010 Diagnosis and Management of Primary Sclerosing Cholangitis [33] | Evaluation and management of bone disease in PBC patients |
| APASL 4 | 2015 Clinical Practice Guidelines on the Management of Hepatitis B [37] | Decline in the bone mineral density in tenofovir disoproxil fumarate treatment |
| Collier et al. | Guidelines on the Management of Osteoporosis Associated with Chronic Liver Disease [9] | Review of the assessment and diagnosis of osteoporosis, the therapeutic agents available, and the way in which they can be used in patients with chronic liver disease to prevent osteoporosis |
| Predominant Changes in Bone Cell Activity in Various Liver Disease | |
|---|---|
| Increased Resorption | Viral hepatitis |
| Transplantation | |
| Corticosteroid therapy | |
| Decreased Formation | Cholestatic liver diseases |
| Iron and copper overload | |
| Risk Factors for a Low Bone Mass and Fragility Fractures |
|---|
| Advanced age |
| Osteoporosis |
| Previous fragility fracture |
| Menopause |
| Male hypogonadism |
| Immobilization or physical inactivity |
| Excess alcohol intake |
| Low body mass index |
| Chronic cholestasis |
| End-stage liver disease |
| Long-term corticosteroid therapy ( >5 mg for more than three months) |
| Immunosuppressive agents |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.M.; Kim, D.J. Bone Diseases in Patients with Chronic Liver Disease. Int. J. Mol. Sci. 2019, 20, 4270. https://doi.org/10.3390/ijms20174270
Jeong HM, Kim DJ. Bone Diseases in Patients with Chronic Liver Disease. International Journal of Molecular Sciences. 2019; 20(17):4270. https://doi.org/10.3390/ijms20174270
Chicago/Turabian StyleJeong, Hae Min, and Dong Joon Kim. 2019. "Bone Diseases in Patients with Chronic Liver Disease" International Journal of Molecular Sciences 20, no. 17: 4270. https://doi.org/10.3390/ijms20174270
APA StyleJeong, H. M., & Kim, D. J. (2019). Bone Diseases in Patients with Chronic Liver Disease. International Journal of Molecular Sciences, 20(17), 4270. https://doi.org/10.3390/ijms20174270