Transcriptome Profiling Provides Insight into the Genes in Carotenoid Biosynthesis during the Mesocarp and Seed Developmental Stages of Avocado (Persea americana)

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Overview of the Morphology and NGST Profiling
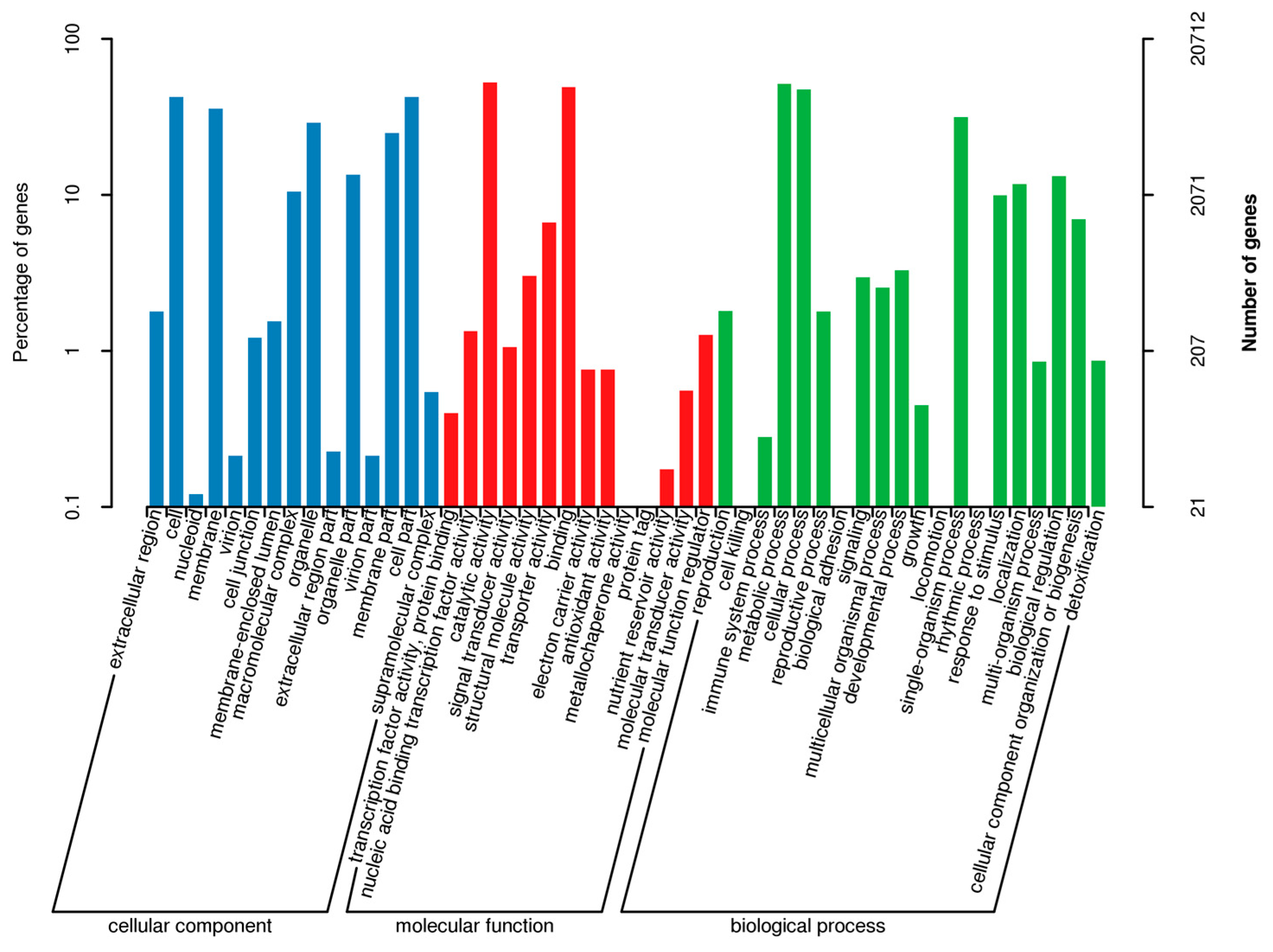
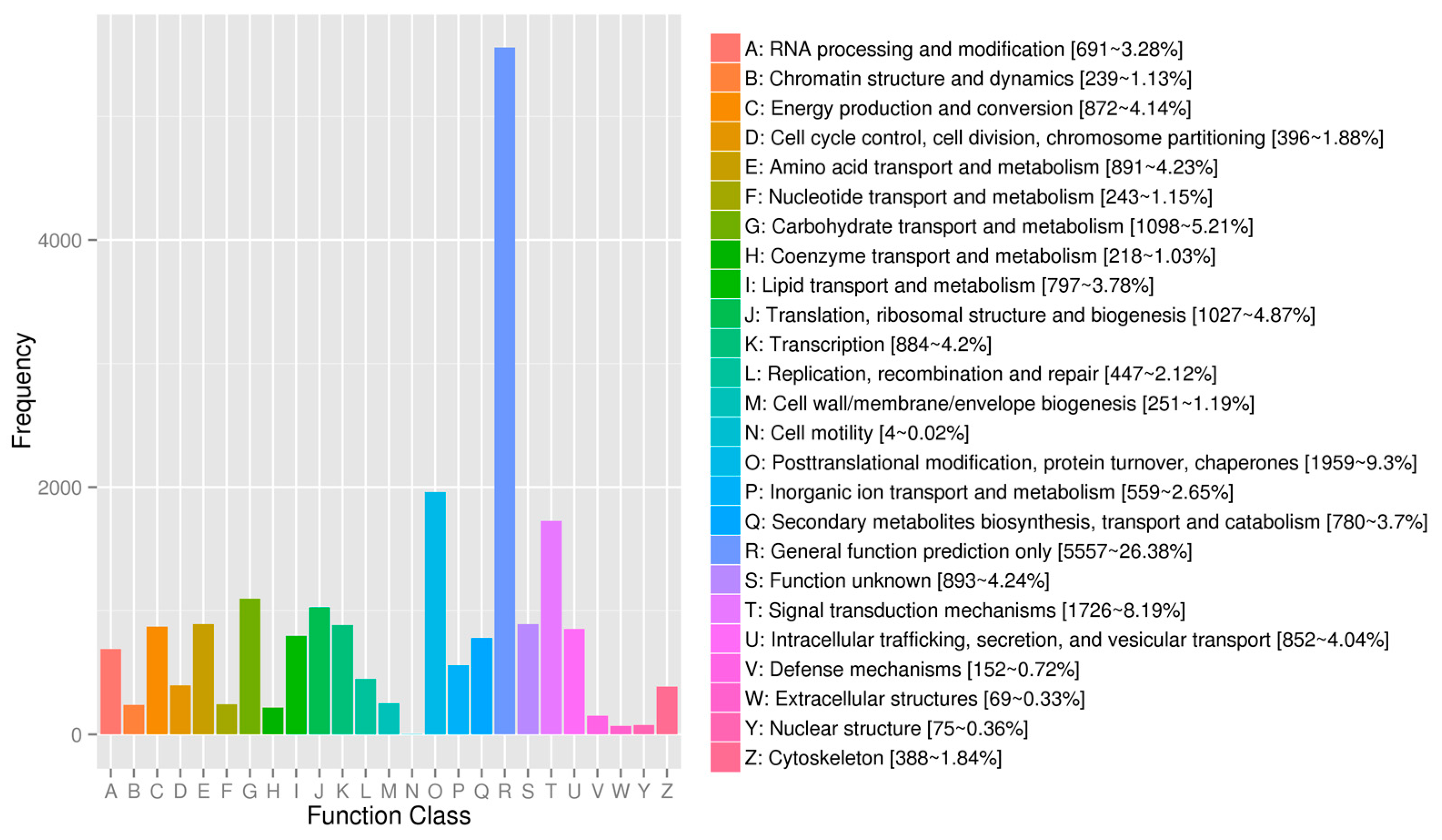
2.2. Annotation and Identification of Unigenes
2.3. Screening of Differentially Expressed Genes during Avocado Mesocarp and Seed Development
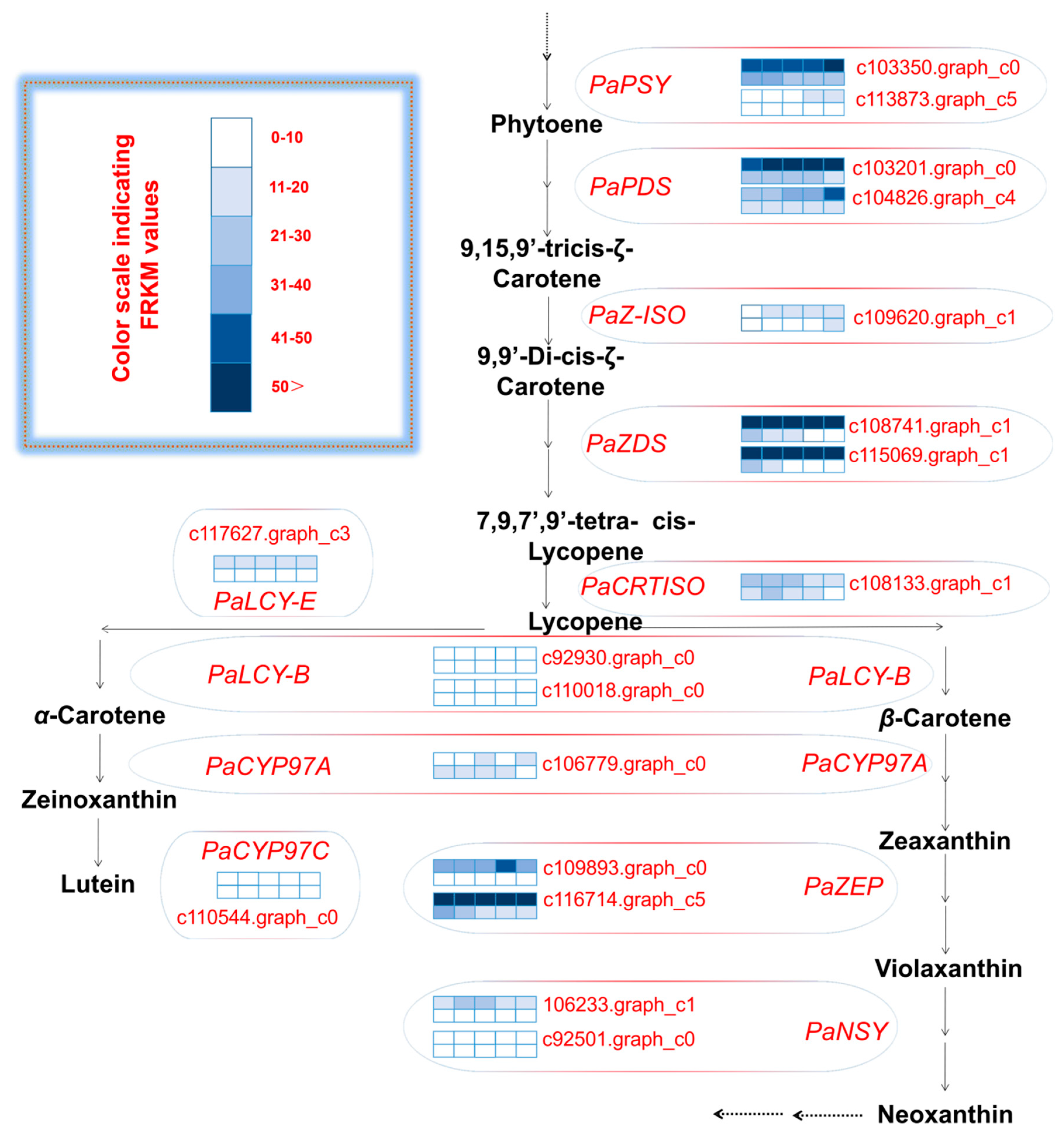
2.4. Differentially Expressed Carotenoid Biosynthetic Genes between the Avocado Mesocarp and Seed
2.5. General Properties of Single-Molecule Long-Reads
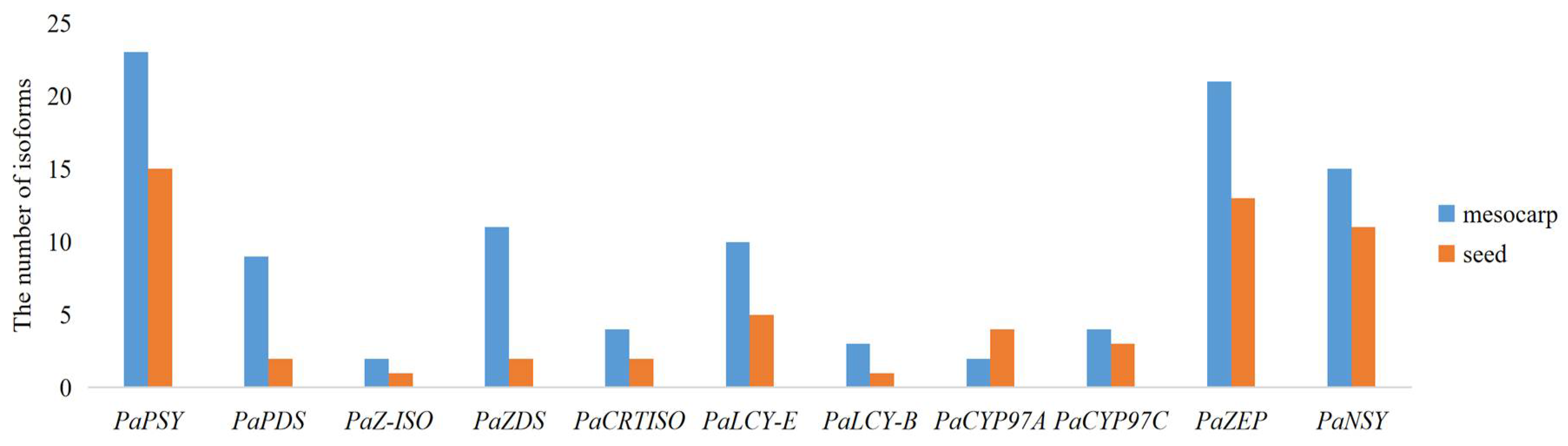
2.6. Isoforms in Carotenoid Biosynthetic Pathway between the Avocado Mesocarp and Seed
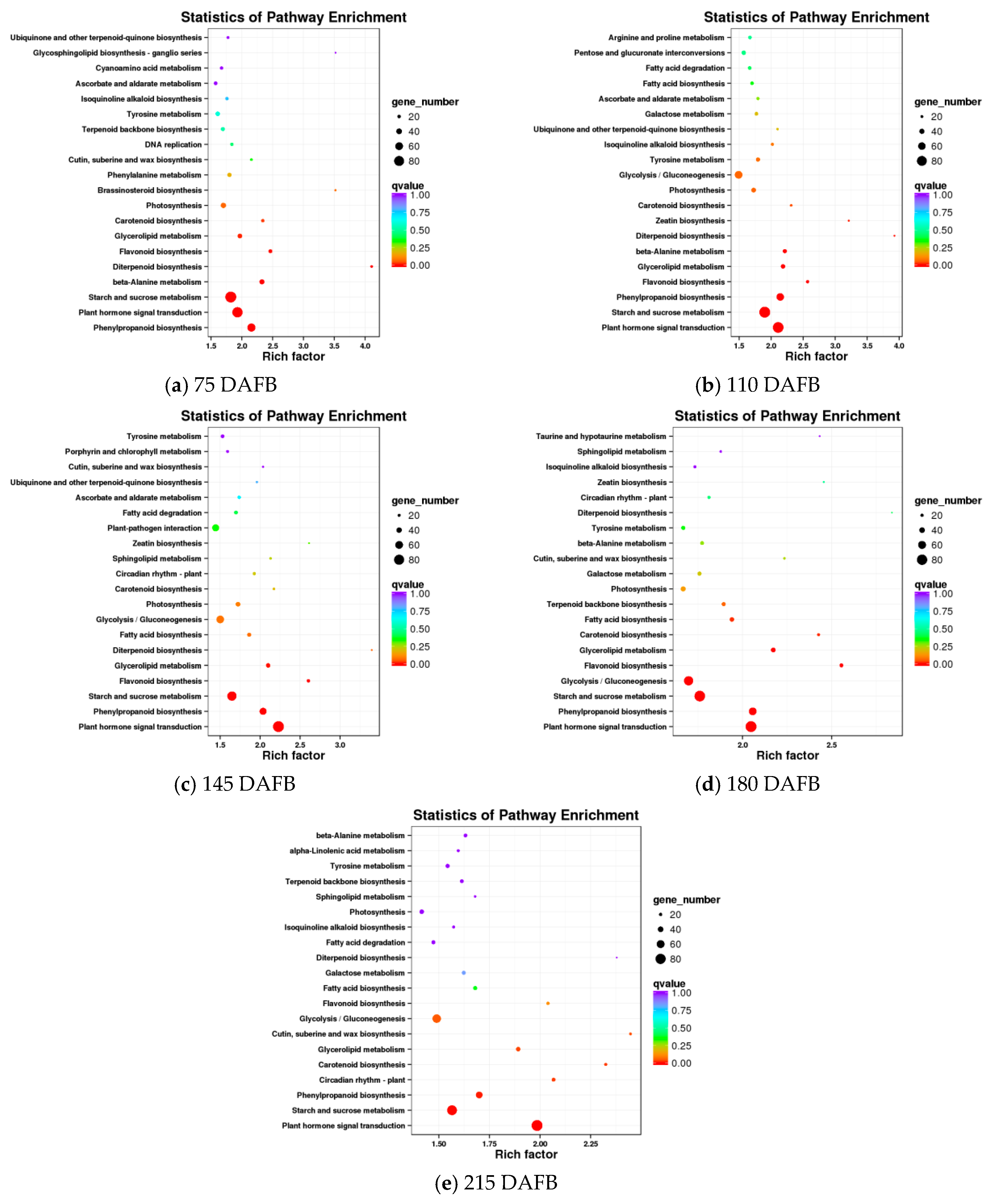
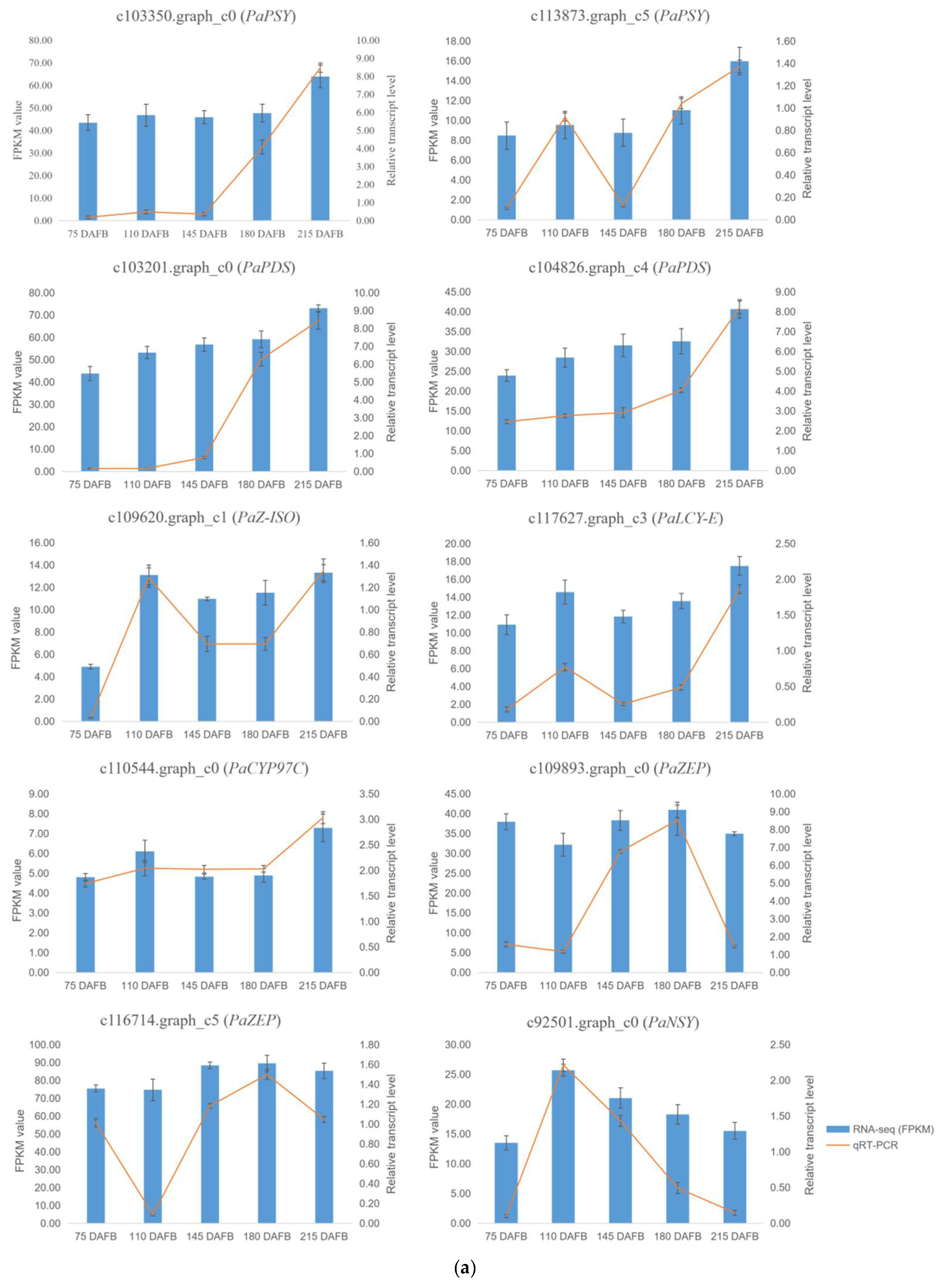
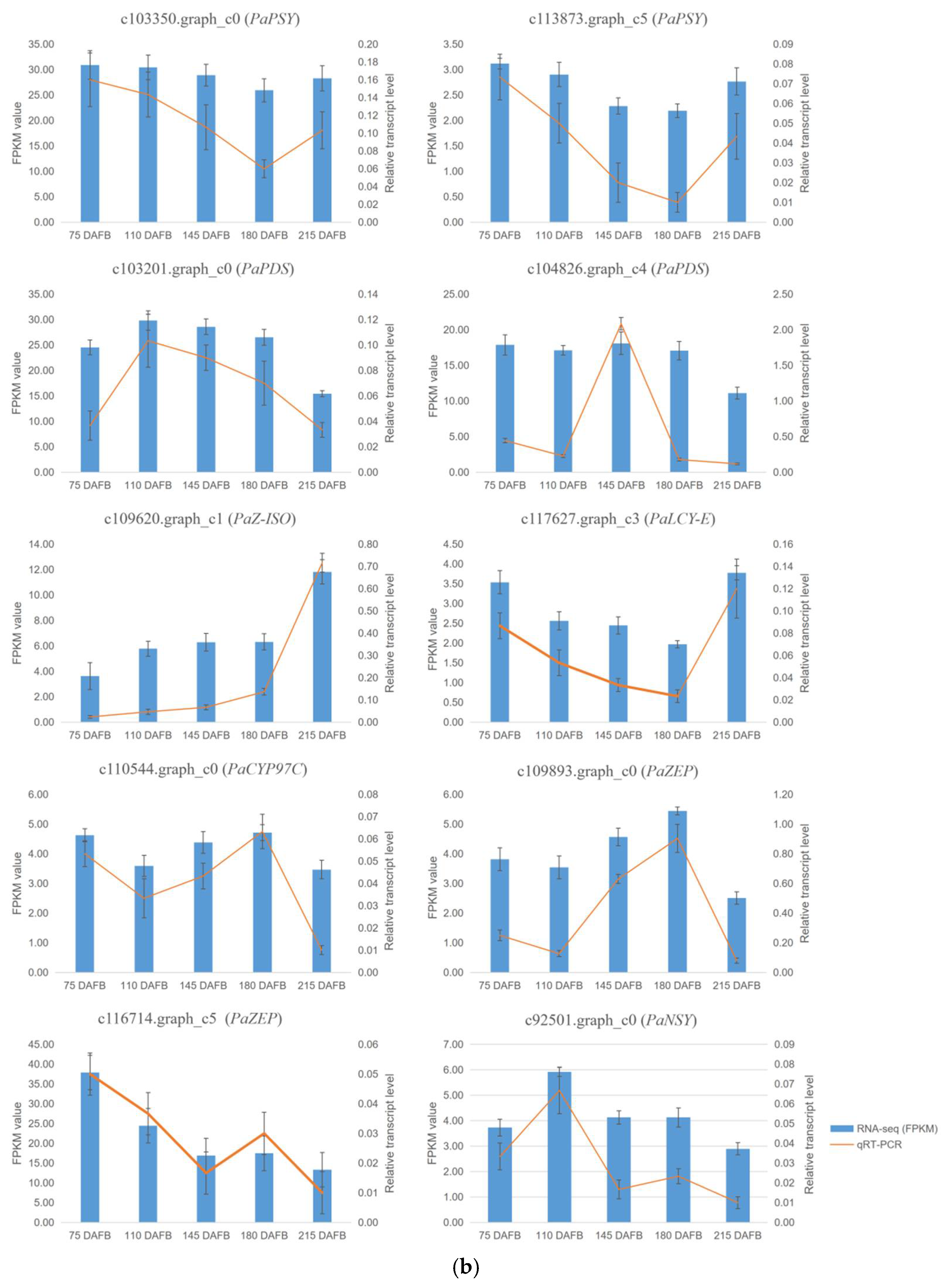
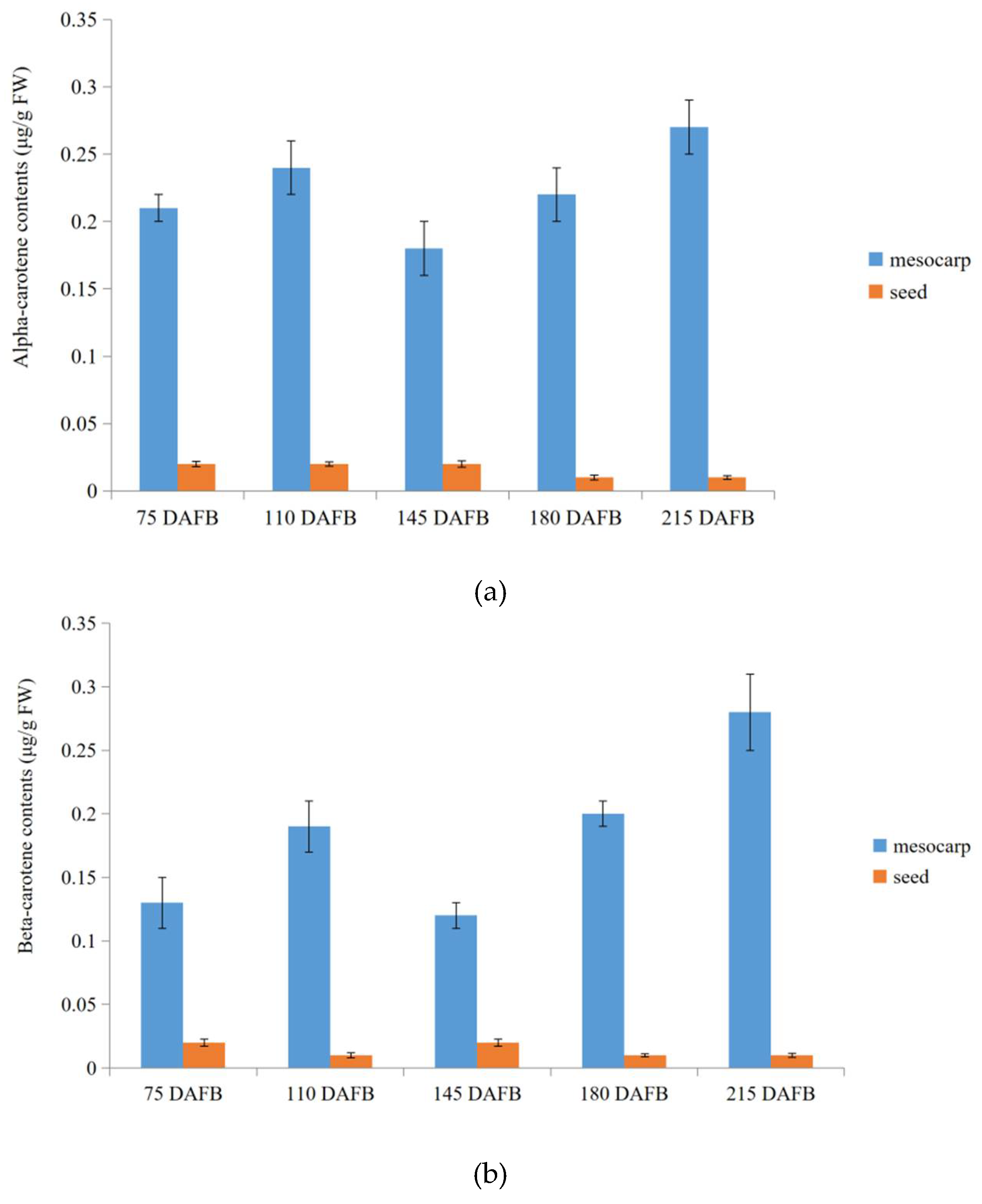
2.7. Verification of Transcriptome Profiling in Carotenoid Biosynthetic Pathway between the Avocado Mesocarp and Seed by Metabolite Profiling via HPLC
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. NGST Sequencing
4.3. Transcriptome Assembly, Annotation, and Coding Sequence Prediction
4.4. Identification of Differentially Expressed Genes
4.5. Validation of Transcripts by Quantitative Real-Time PCR
4.6. SMRT Sequencing
4.7. Quality Filtering and Correction of PacBio Long-Reads
4.8. Analysis of Alpha- and Beta-Carotenes by HPLC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gross-German, E.; Viruel, M.A. Molecular characterization of avocado germplasm with a new set of SSR and EST-SSR markers: Genetic diversity, population structure, and identification of race-specific markers in a group of cultivated genotypes. Tree Genet. Genomes 2013, 9, 539–555. [Google Scholar] [CrossRef]
- Galindo-Tovar, M.E.; Ogata-Aguilar, N.; Arzate-Fernandez, A.M. Some aspects of avocado (Persea americana Mill.) diversity and domestication in Mesoamerica. Genet. Resour. Crop Evol. 2008, 55, 441–450. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, T.; Wu, B.; Tan, L.; Ma, F.; Zou, M.; Chen, H.; Pei, J.; Liu, Y.; Chen, Z.; et al. Genome-Wide Assessment of Avocado Germplasm Determined from Specific Length Amplified Fragment Sequencing and Transcriptomes: Population Structure, Genetic Diversity, Identification, and Application of Race-Specific Markers. Genes 2019, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, B.; Wolstenholme, B.N.; Whiley, A.W. The Avocado: Botany, Production and Uses, 2nd ed.; CPI Group Ltd.: Croydon, UK, 2012. [Google Scholar]
- Dreher, M.L.; Davenport, A.J. Hass Avocado Composition and Potential Health Effects. Crit. Rev. Food Sci. Nutr. 2013, 53, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Galvão, M.D.S.; Narain, N.; Nigam, N. Influence of different cultivars on oil quality and chemical characteristics of avocado fruit. Food Sci. Technol. 2014, 34, 539–546. [Google Scholar] [CrossRef]
- Ge, Y.; Si, X.; Cao, J.; Zhou, Z.; Wang, W.; Ma, W. Morphological Characteristics, Nutritional Quality, and Bioactive Constituents in Fruits of Two Avocado (Persea americana) Varieties from Hainan Province, China. J. Agric. Sci. 2017, 9, 8–17. [Google Scholar] [CrossRef]
- Joung, J.G.; Chung, M.Y.; Tieman, D.; Lee, J.M.; Joung, J.; McQuinn, R.; Chung, M.; Fei, Z.; Klee, H.; Giovannoni, J. Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J. 2012, 70, 191–204. [Google Scholar]
- Li, J.W.; Ma, J.; Feng, K.; Liu, J.X.; Que, F.; Xiong, A.S. Carotenoid Accumulation and Distinct Transcript Profiling of Structural Genes Involved in Carotenoid Biosynthesis in Celery. Plant Mol. Biol. Rep. 2018, 36, 663–674. [Google Scholar] [CrossRef]
- Goff, S.A.; Klee, H.J. Plant Volatile Compounds: Sensory Cues for Health and Nutritional Value? Scinece 2006, 311, 815–819. [Google Scholar] [CrossRef]
- Walter, M.H.; Floss, D.S.; Strack, D. Apocarotenoids: Hormones, mycorrhizal metabolites and aroma volatiles. Planta 2010, 232, 1–17. [Google Scholar] [CrossRef]
- Nisar, N.; Li, L.; Lu, S.; Khin, N.C.; Pogson, B.J. Carotenoid Metabolism in Plants. Mol. Plant 2015, 8, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Britton, G. Structure and properties of carotenoids in relation to function. FASEB J. 1995, 9, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, J.; Nageswaran, D.; Li, L. Carotenoid metabolism and regulation in horticultural crops. Hortic. Res. 2015, 2, 15036. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, H.; Wang, Y.; Zong, J.; Chen, J.; Li, D.D.; Li, L.; Wang, J.J.; Liu, J.X. High-throughput SSR marker development and its application in a centipedegrass (Eremochloa ophiuroides (Munro) Hack.) genetic diversity analysis. PLoS ONE 2018, 13, e0202605. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Jiang, X.; Liu, Q.; Liu, Q.; Wang, K. De Novo Transcriptomic Analysis and Development of EST–SSRs for Styrax japonicus. Forests 2018, 9, 748. [Google Scholar] [CrossRef]
- Liu, F.M.; Hong, Z.; Yang, Z.J.; Zhang, N.N.; Liu, X.J.; Xu, D.P. De Novo transcriptome analysis of Dalbergia odorifera T. Chen (Fabaceae) and transferability of SSR markers developed from the transcriptome. Forests 2019, 10, 98. [Google Scholar] [CrossRef]
- Ma, S.; Dong, W.; Lyu, T.; Lyu, Y. An RNA Sequencing Transcriptome Analysis and Development of EST-SSR Markers in Chinese Hawthorn through Illumina Sequencing. Forests 2019, 10, 82. [Google Scholar] [CrossRef]
- Penin, A.A.; Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D. Comparative Analysis of Developmental Transcriptome Maps of Arabidopsis thaliana and Solanum lycopersicum. Genes 2019, 10, 50. [Google Scholar] [CrossRef]
- Weisberg, A.J.; Kim, G.; Westwood, J.H.; Jelesko, J.G. Sequencing and De Novo Assembly of the Toxicodendron radicans (Poison Ivy) Transcriptome. Genes 2017, 8, 317. [Google Scholar] [CrossRef]
- Hyun, T.K.; Rim, Y.; Jang, H.J.; Kim, C.H.; Park, J.; Kumar, R.; Lee, S.; Kim, B.C.; Bhak, J.; Nguyen-Quoc, B.; et al. De novo transcriptome sequencing of Momordica cochinchinensis to identify genes involved in the carotenoid biosynthesis. Plant Mol. Biol. 2012, 79, 413–427. [Google Scholar] [CrossRef]
- Tian, H.M.; Fang, L.; Zhang, Q.A.; Wang, M.X.; Wang, Y.; Jia, L. Transcriptome analysis of carotenoid biosynthesis in the Brassica campestris L. subsp. chinensis var. rosularis Tsen. Sci. Hortic. 2018, 235, 116–123. [Google Scholar] [CrossRef]
- He, Y.; Ma, Y.; Du, Y.; Shen, S. Differential gene expression for carotenoid biosynthesis in a green alga Ulva prolifera based on transcriptome analysis. BMC Genom. 2018, 19, 916. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, J.; Xu, Z.; Wang, F.; Xiong, A. Transcriptome profiling of genes involving in carotenoid biosynthesis and accumulation between leaf and root of carrot (Daucus carota L.). Acta Biochim. Biophys. Sin. 2018, 50, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Machaj, G.; Bostan, H.; Macko-Podgórni, A.; Iorizzo, M.; Grzebelus, D. Comparative Transcriptomics of Root Development in Wild and Cultivated Carrots. Genes 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Sun, W.; Zou, X.; Liu, B.; Huang, W.; Chen, Z.; Li, Y.; Qiu, M.-Y.; Liu, Z.-J.; Mao, Y.; et al. Sequencing of Euscaphis konishii Endocarp Transcriptome Points to Molecular Mechanisms of Endocarp Coloration. Int. J. Mol. Sci. 2018, 19, 3209. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Feng, S.; Liu, Y.; Zhao, L.; Tian, L.; Hu, Y.; Yang, T.; Wei, A. Single-Molecule Long-Read Sequencing of Zanthoxylum bungeanum Maxim. Transcriptome: Identification of Aroma-Related Genes. Forest 2018, 9, 765. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Yuan, J.B.; Li, S.F.; Jia, S.Q.; Han, L.B.; Xu, L.X. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Blow, M.; Sreedasyam, A.; Kuo, R.C.; Ramamoorthy, G.K.; Torres-Jerez, I.; Li, G.; Wang, M.; Dilworth, D.; Barry, K.; et al. Revealing the transcriptomic complexity of switchgrass by PacBio long-read sequencing. Biotechnol. Biofuels 2018, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Yuan, J.; Guo, T.; Xu, L.; Mu, Z.; Han, L. Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing. Plant Mol. Biol. 2019, 99, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Roy, N.S.; Lee, I.H.; Choi, A.Y.; Choi, B.S.; Yu, Y.S.; Park, N.I.; Park, K.C.; Kim, S.; Yang, H.S.; et al. Genome-wide transcriptome profiling of the medicinal plant Zanthoxylum planispinum using a single-molecule direct RNA sequencing approach. Genomics 2019, 111, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, J.; Wang, X.; Wei, Z. Full-length RNA sequencing reveals unique transcriptome composition in bermudagrass. Plant Physiol. Biochem. 2018, 132, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhu, J.; Zhao, S.; Hou, Y.; Li, F.; Tai, Y.; Wan, X.; Wei, C. Transcriptome Profiling Using Single-Molecule Direct RNA Sequencing Approach for In-depth Understanding of Genes in Secondary Metabolism Pathways of Camellia sinensis. Front. Plant Sci. 2017, 8, 1205. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zheng, H.; Yan, Z.; Liao, D.; Li, C.; Zhou, J.; Liao, H. Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response. Int. J. Mol. Sci. 2018, 19, 2476. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Li, N.; Niu, B.; Liu, Y.; You, D.; Jiang, D.; Ruan, C.; Qin, Z.; Song, T.; Wang, W. De novo transcriptome analysis of Bagarius yarrelli (Siluriformes: Sisoridae) and the search for potential SSR markers using RNA-Seq. PLoS ONE 2018, 13, e0190343. [Google Scholar] [CrossRef]
- Ibarra-Laclette, E.; Méndez-Bravo, A.; Pérez-Torres, C.A.; Albert, V.A.; Mockaitis, K.; Kilaru, A.; López-Gómez, R.; Cervantes-Luevano, J.I.; Herrera-Estrella, L. Deep sequencing of the Mexican avocado transcriptome, an ancient angiosperm with a high content of fatty acids. BMC Genom. 2015, 16, 599. [Google Scholar] [CrossRef]
- Kilaru, A.; Cao, X.; Dabbs, P.B.; Sung, H.-J.; Rahman, M.M.; Thrower, N.; Zynda, G.; Podicheti, R.; Ibarra-Laclette, E.; Herrera-Estrella, L.; et al. Oil biosynthesis in a basal angiosperm: Transcriptome analysis of Persea Americana mesocarp. BMC Plant Biol. 2015, 15, 203. [Google Scholar] [CrossRef]
- Vergara-Pulgar, C.; Rothkegel, K.; González-Agüero, M.; Pedreschi, R.; Campos-Vargas, R.; Defilippi, B.G.; Meneses, C. De novo assembly of Persea americana cv. ‘Hass’ transcriptome during fruit development. BMC Genom. 2019, 20, 108. [Google Scholar] [CrossRef]
- Ge, Y.; Tan, L.; Wu, B.; Wang, T.; Zhang, T.; Chen, H.; Zou, M.; Ma, F.; Xu, Z.; Zhan, R. Transcriptome Sequencing of Different Avocado Ecotypes: De novo Transcriptome Assembly, Annotation, Identification and Validation of EST-SSR Markers. Forests 2019, 10, 411. [Google Scholar] [CrossRef]
- Wisutiamonkul, A.; Ampomah-Dwamena, C.; Allan, A.C.; Ketsa, S. Carotenoid accumulation in durian (Durio zibethinus) fruit is affected by ethylene via modulation of carotenoid pathway gene expression. Plant Physiol. Biochem. 2017, 115, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Bastiaanse, H.; Zinkgraf, M.; Canning, C.; Tsai, H.; Lieberman, M.; Comai, L.; Henry, I.; Groover, A. A comprehensive genomic scan reveals gene dosage balance impacts on quantitative traits in Populus trees. Proc. Natl. Acad. Sci. USA 2019, 116, 13690–13699. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Bhadra, U.; Bhadra, M.P.; Auger, D.L. Dosage-Dependent Gene Regulation in Multicellular Eukaryotes: Implications for Dosage Compensation, Aneuploid Syndromes, and Quantitative Traits. Dev. Biol. 2001, 234, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Förster, S.; Schumann, E.; Baumann, M.; Weber, W.E.; Pillen, K. Copy number variation of chromosome 5A and its association with Q gene expression, morphological aberrations, and agronomic performance of winter wheat cultivars. Theor. Appl. Genet. 2013, 126, 3049–3063. [Google Scholar] [CrossRef] [PubMed]
- Bramley, P.M. Regulation of carotenoid formation during tomato fruit ripening and development. J. Exp. Bot. 2002, 53, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Devitt, L.C.; Fanning, K.; Dietzgen, R.G.; Holton, T.A. Isolation and functional characterization of a lycopene β-cyclase gene that controls fruit colour of papaya (Carica papaya L.). J. Exp. Bot. 2010, 61, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Ampomah-Dwamena, C.; McGhie, T.; Wibisono, R.; Montefiori, M.; Hellens, R.P.; Allan, A.C. The kiwifruit lycopene beta-cyclase plays a significant role in carotenoid accumulation in fruit. J. Exp. Bot. 2009, 60, 3765–3779. [Google Scholar] [CrossRef]
- Kato, M.; Ikoma, Y.; Matsumoto, H.; Sugiura, M.; Hyodo, H.; Yano, M. Accumulation of carotenoids and expression of carotenoid biosynthetic genes during maturation in citrus fruit. Plant Physiol. 2004, 134, 824–837. [Google Scholar] [CrossRef]
- Hornero-Méndez, D.; Gómez-Ladrón, D.G.R.; Mínguez-Mosquera, M.I. Carotenoid biosynthesis changes in five red pepper (Capsicum annuum L.) cultivars during ripening. Cultivar selection for breeding. J. Agric. Food Chem. 2000, 48, 3857–3864. [Google Scholar] [CrossRef]
- Ducreux, L.J.; Morris, W.L.; Hedley, P.E.; Shepherd, T.; Davies, H.V.; Millam, S.; Taylor, M.A. Metabolic engineering of high carotenoid potato tubers containing enhanced levels of beta-carotene and lutein. J. Exp. Bot. 2005, 56, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Welsch, R.; Arango, J.; Bär, C.; Salazar, B.; Al-Babili, S.; Beltrán, J.; Chavarriaga, P.; Ceballos, H.; Tohme, J.; Beyer, P. Provitamin A Accumulation in Cassava (Manihot esculenta) Roots Driven by a Single Nucleotide Polymorphism in a Phytoene Synthase Gene. Plant Cell 2010, 22, 3348–3356. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.Y.; Zhang, Y.; Wang, Y.; Wang, D.; Lee, R.P.; Gao, K.; Byrns, R.; Heber, D. California Hass Avocado: Profiling of Carotenoids, tocopherol, fatty acid, and fat content during maturation and from different growing areas. J. Agric. Food Chem. 2009, 57, 10408–10413. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Functional Protein Name | Enzyme Commission Number | Unigene ID |
|---|---|---|---|
| PaPSY | 15-cis-phytoene synthase | 2.5.1.32 | c103350.graph_c0, c113873.graph_c5 |
| PaPDS | Phytoene desaturase | 1.3.5.5 | c103201.graph_c0, c104826.graph_c4 |
| PaZ-ISO | 15-cis-ζ-carotene isomerase | 5.2.1.12 | c109620.graph_c1 |
| PaZDS | ζ-carotene desaturase | 1.3.5.6 | c108741.graph_c1, c115069.graph_c3 |
| PaCRTISO | Carotenoid isomerase | 5.2.1.13 | c108133.graph_c1 |
| PaLCY-E | Lycopene ε-cyclase | 5.5.1.18 | c117627.graph_c3 |
| PaLCY-B | Lycopene β-cyclase | 5.5.1.19 | c92930.graph_c0, c110018.graph_c0 |
| PaCYP97A | P450 β-ring carotene hydroxylase | 1.14.13.129 | c106779.graph_c0 |
| PaCYP97C | P450 ε-ring carotene hydroxylase | 1.14.99.45 | c110544.graph_c0 |
| PaZEP | Zeaxanthin epoxidase | 1.14.15.21 | c109893.graph_c0, c116714.graph_c5 |
| PaNSY | Neoxanthin synthase | 5.3.99.9 | c106233.graph_c1, c92501.graph_c0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, Y.; Cheng, Z.; Si, X.; Ma, W.; Tan, L.; Zang, X.; Wu, B.; Xu, Z.; Wang, N.; Zhou, Z.; et al. Transcriptome Profiling Provides Insight into the Genes in Carotenoid Biosynthesis during the Mesocarp and Seed Developmental Stages of Avocado (Persea americana). Int. J. Mol. Sci. 2019, 20, 4117. https://doi.org/10.3390/ijms20174117
Ge Y, Cheng Z, Si X, Ma W, Tan L, Zang X, Wu B, Xu Z, Wang N, Zhou Z, et al. Transcriptome Profiling Provides Insight into the Genes in Carotenoid Biosynthesis during the Mesocarp and Seed Developmental Stages of Avocado (Persea americana). International Journal of Molecular Sciences. 2019; 20(17):4117. https://doi.org/10.3390/ijms20174117
Chicago/Turabian StyleGe, Yu, Zhihao Cheng, Xiongyuan Si, Weihong Ma, Lin Tan, Xiaoping Zang, Bin Wu, Zining Xu, Nan Wang, Zhaoxi Zhou, and et al. 2019. "Transcriptome Profiling Provides Insight into the Genes in Carotenoid Biosynthesis during the Mesocarp and Seed Developmental Stages of Avocado (Persea americana)" International Journal of Molecular Sciences 20, no. 17: 4117. https://doi.org/10.3390/ijms20174117
APA StyleGe, Y., Cheng, Z., Si, X., Ma, W., Tan, L., Zang, X., Wu, B., Xu, Z., Wang, N., Zhou, Z., Lin, X., Dong, X., & Zhan, R. (2019). Transcriptome Profiling Provides Insight into the Genes in Carotenoid Biosynthesis during the Mesocarp and Seed Developmental Stages of Avocado (Persea americana). International Journal of Molecular Sciences, 20(17), 4117. https://doi.org/10.3390/ijms20174117