Antagonistic Effects of IL-4 on IL-17A-Mediated Enhancement of Epidermal Tight Junction Function

 , , and
, , and 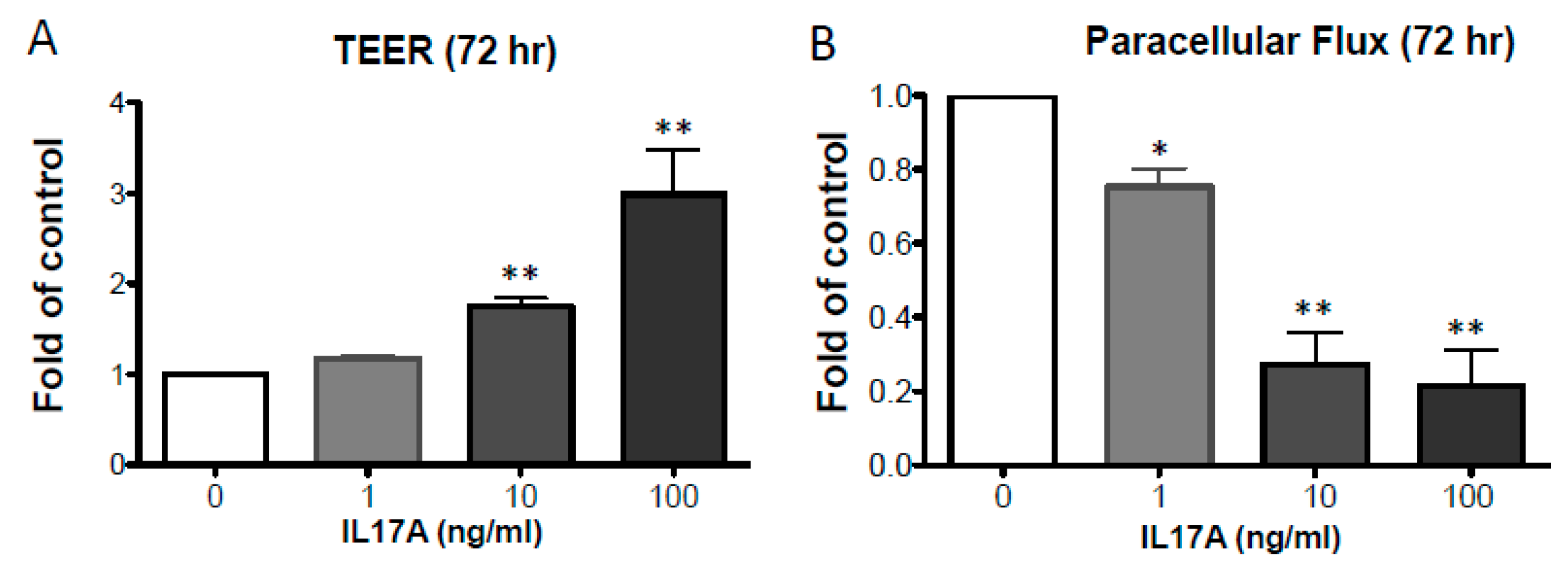{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. IL-17A Enhances Epidermal TJ Barrier Integrity
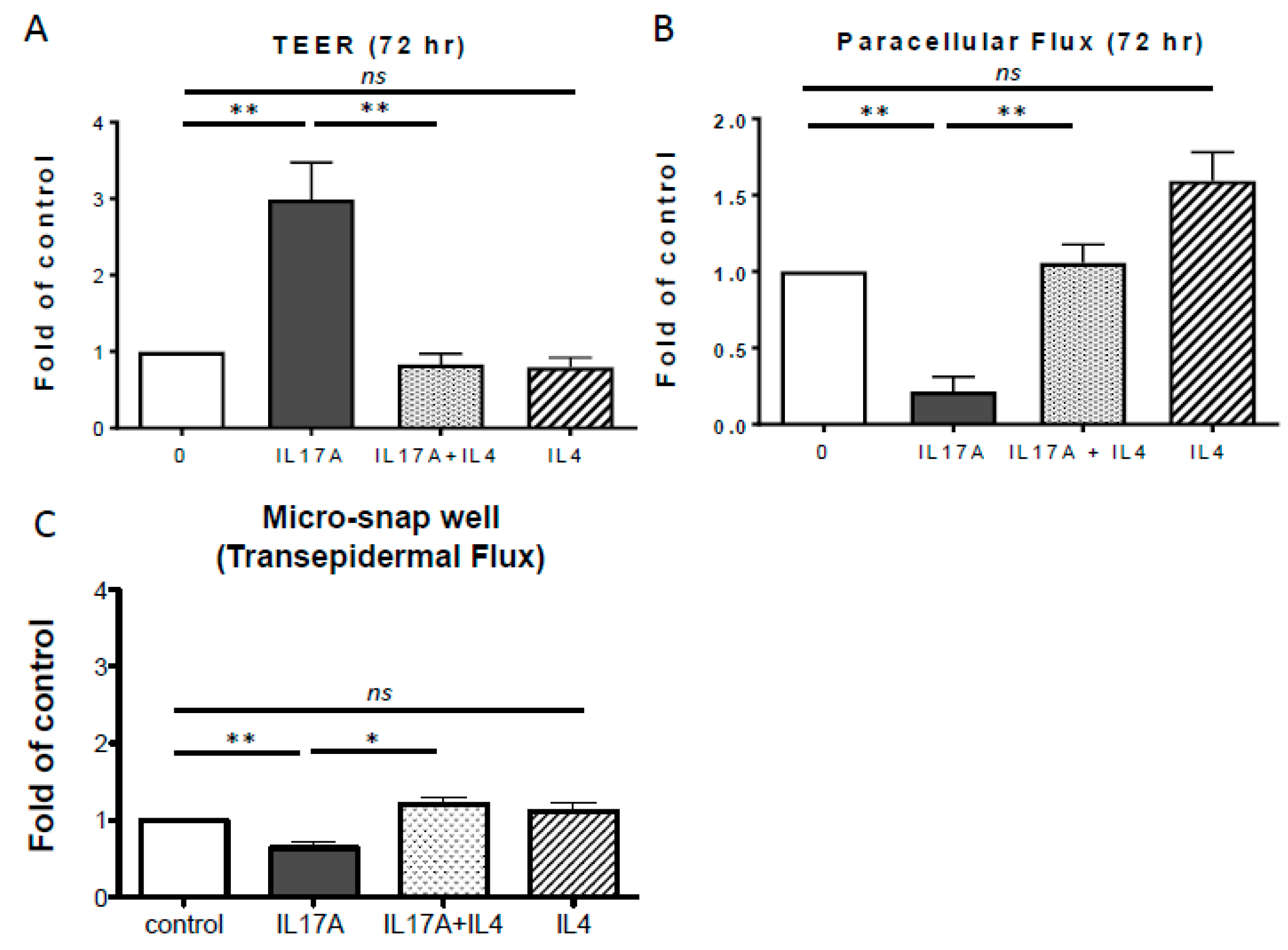
2.2. IL-4 Inhibits IL-17A-Mediated TJ Barrier Enhancement
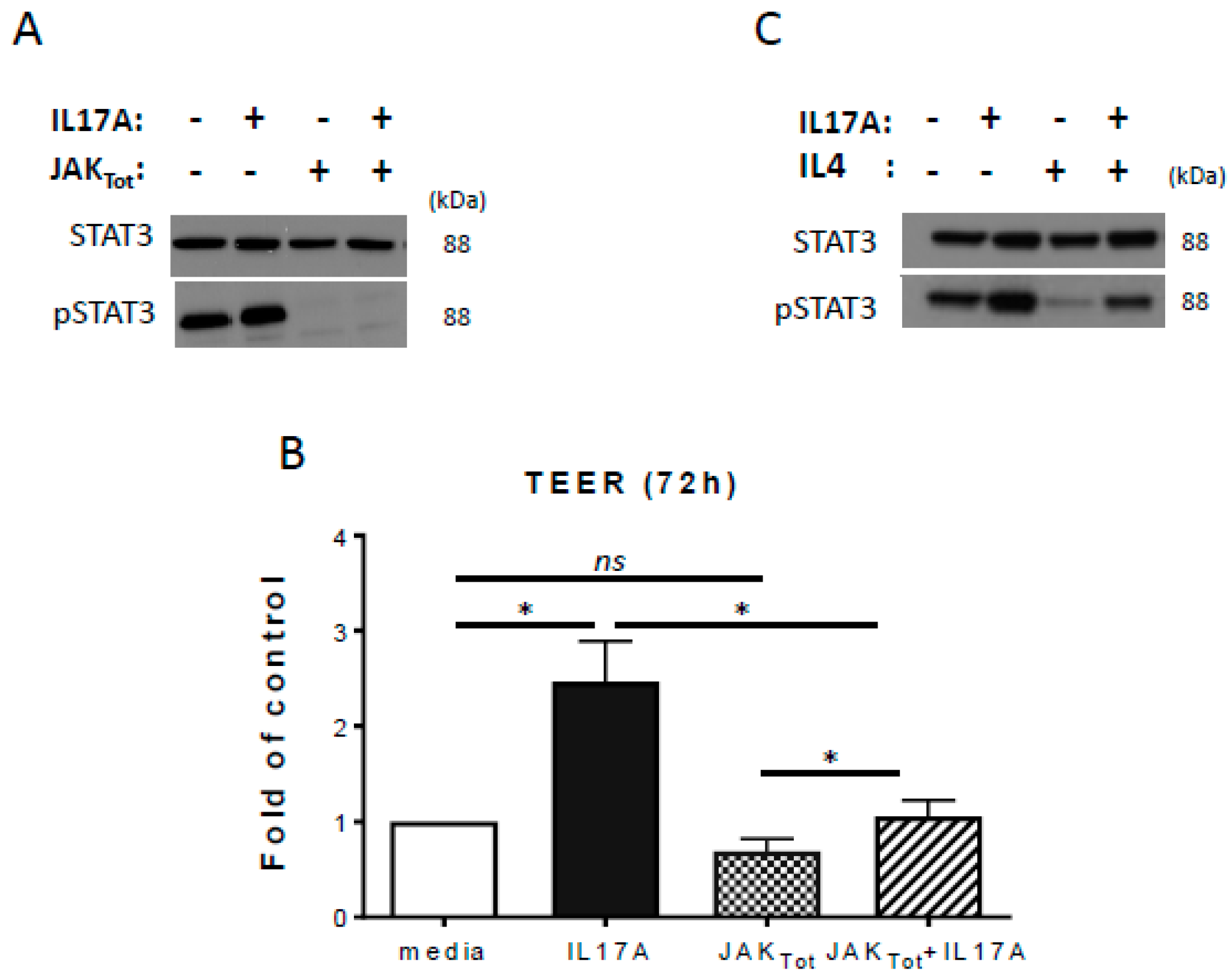
2.3. Inhibition of STAT3 Activation Reduces IL-17A-Induced TEER
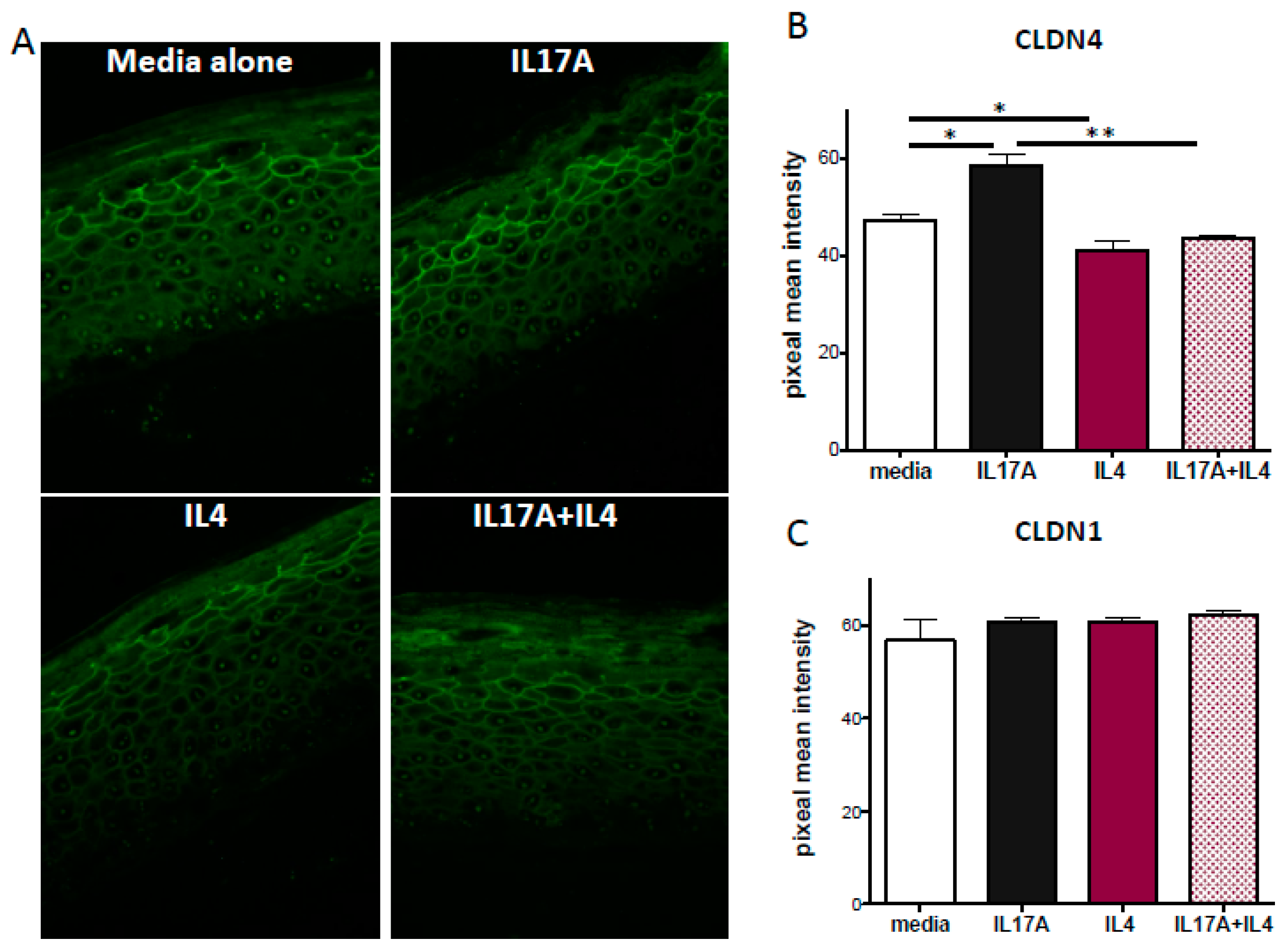
2.4. IL-17A Enhances Claudin-4 Expression
3. Discussion
4. Methods
4.1. Isolation and Culture of Primary Human Foreskin Keratinocytes
4.2. Transepithelial Electric Resistance (TEER) and Paracellular Flux
4.3. Immunoblotting
4.4. Immunostaining
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, T.C.; Sanyal, R.D.; Pavel, A.B.; Glickman, J.; Zheng, X.; Xu, H.; Cho, Y.T.; Tsai, T.F.; Wen, H.C.; Peng, X.; et al. Atopic dermatitis in Chinese patients shows TH2/TH17 skewing with psoriasiform features. J. Allergy Clin. Immunol. 2018, 142, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, R.D.; Pavel, A.B.; Glickman, J.; Chan, T.C.; Zheng, X.; Zhang, N.; Cueto, I.; Peng, X.; Estrada, Y.; Fuentes-Duculan, J.; et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann. Allergy Asthma Immunol. 2019, 122, 99–110. [Google Scholar]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar]
- Yuki, T.; Tobiishi, M.; Kusaka-Kikushima, A.; Ota, Y.; Tokura, Y. Impaired Tight Junctions in Atopic Dermatitis Skin and in a Skin-Equivalent Model Treated with Interleukin-17. PLoS ONE 2016, 11, e0161759. [Google Scholar] [CrossRef] [PubMed]
- Batista, D.I.S.; Perez, L.; Orfali, R.L.; Zaniboni, M.C.; Samorano, L.P.; Pereira, N.V.; Sotto, M.N.; Ishizaki, A.S.; Oliveira, L.M.S.; Sato, M.N.; et al. Profile of skin barrier proteins (filaggrin, claudins 1 and 4) and Th1/Th2/Th17 cytokines in adults with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1091–1095. [Google Scholar] [PubMed]
- Kast, J.I.; Wanke, K.; Soyka, M.B.; Wawrzyniak, P.; Akdis, D.; Kingo, K.; Rebane, A.; Akdis, C.A. The broad spectrum of interepithelial junctions in skin and lung. J. Allergy Clin. Immunol. 2012, 130, 544–547. [Google Scholar] [CrossRef]
- Kirschner, N.; Poetzl, C.; von den Driesch, P.; Wladykowski, E.; Moll, I.; Behne, M.J.; Brandner, J.M. Alteration of tight junction proteins is an early event in psoriasis: Putative involvement of proinflammatory cytokines. Am. J. Pathol. 2009, 175, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Taylor, S.; Ogg, G.S. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol. 2011, 165, 492–498. [Google Scholar]
- Hvid, M.; Vestergaard, C.; Kemp, K.; Christensen, G.B.; Deleuran, B.; Deleuran, M. IL-25 in atopic dermatitis: A possible link between inflammation and skin barrier dysfunction? J. Investig. Dermatol. 2011, 131, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fairchild, H.R.; Kim, B.E.; Bin, L.; Boguniewicz, M.; Redzic, J.S.; Hansen, K.C.; Leung, D.Y. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J. Investig. Dermatol. 2008, 128, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Floudas, A.; Saunders, S.P.; Moran, T.; Schwartz, C.; Hams, E.; Fitzgerald, D.C.; Johnston, J.A.; Ogg, G.S.; McKenzie, A.N.; Walsh, P.T.; et al. IL-17 Receptor A Maintains and Protects the Skin Barrier to Prevent Allergic Skin Inflammation. J. Immunol. 2017, 199, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, P.; Henehan, M.; De Benedetto, A. Activation of protease-activated receptor 2 leads to impairment of keratinocyte tight junction integrity. J. Allergy Clin. Immunol. 2018, 142, 281–284. [Google Scholar] [CrossRef] [PubMed]
- El Asmar, R.; Panigrahi, P.; Bamford, P.; Berti, I.; Not, T.; Coppa, G.V.; Catassi, C.; Fasano, A. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology 2002, 123, 1607–1615. [Google Scholar] [CrossRef]
- Kuo, I.H.; Carpenter-Mendini, A.; Yoshida, T.; McGirt, L.Y.; Ivanov, A.I.; Barnes, K.C.; Gallo, R.L.; Borkowski, A.W.; Yamasaki, K.; Leung, D.Y.; et al. Activation of epidermal toll-like receptor 2 enhances tight junction function: Implications for atopic dermatitis and skin barrier repair. J. Investig. Dermatol. 2013, 133, 988–998. [Google Scholar] [PubMed]
- Eyerich, K.; Pennino, D.; Scarponi, C.; Foerster, S.; Nasorri, F.; Behrendt, H.; Ring, J.; Traidl-Hoffmann, C.; Albanesi, C.; Cavani, A. IL-17 in atopic eczema: Linking allergen-specific adaptive and microbial-triggered innate immune response. J. Allergy Clin. Immunol. 2009, 123, 59–66. [Google Scholar] [CrossRef]
- Nograles, K.E.; Suárez-Fariñas, M.; Shemer, A.; Fuentes-Duculan, J.; Chiricozzi, A.; Cardinale, I.; Zaba, L.C.; Kikuchi, T.; Ramon, M.; Bergman, R.; et al. Atopic dermatitis (AD) keratinocytes exhibit normal Th17 cytokine responses. J. Investig. Dermatol. 2010, 125, 744–746. [Google Scholar]
- Bai, B.; Yamamoto, K.; Sato, H.; Sugiura, H.; Tanaka, T. Complex regulation of S100A8 by IL-17, dexamethasone, IL-4 and IL-13 in HaCat cells (human keratinocyte cell line). J. Dermatol. Sci. 2007, 47, 259–262. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Dermatol. 2012, 21, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Zaba, L.C.; Guttman-Yassky, E.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Cardinale, I.; Khatcherian, A.; Gonzalez, J.; Pierson, K.C.; White, T.R.; et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol. 2008, 159, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Kao, C.Y.; Wachi, S.; Thai, P.; Ryu, J.; Wu, R. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J. Immunol. 2007, 179, 6504–6513. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, D.C.; Boswell, M.G.; Zhou, W.; Huckabee, M.M.; Goleniewska, K.; Sevin, C.M.; Hershey, G.K.K.; Kolls, J.K.; Peebles, R.S., Jr. Human TH17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J. Allergy Clin. Immunol. 2011, 127, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Itami, S.; Takeda, K.; Tarutani, M.; Yamaguchi, Y.; Miura, H.; Yoshikawa, K.; Akira, S.; Takeda, J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999, 18, 4657–4668. [Google Scholar] [CrossRef]
- Wang, Z.; Li, R.; Tan, J.; Peng, L.; Wang, P.; Liu, J.; Xiong, H.; Jiang, B.; Chen, Y. Syndecan-1 Acts in Synergy with Tight Junction Through Stat3 Signaling to Maintain Intestinal Mucosal Barrier and Prevent Bacterial Translocation. Inflamm. Bowel Dis. 2015, 21, 1894–1907. [Google Scholar] [CrossRef]
- Yun, J.H.; Park, S.W.; Kim, K.J.; Bae, J.S.; Lee, E.H.; Paek, S.H.; Kim, S.U.; Ye, S.; Kim, J.H.; Cho, C.H. Endothelial STAT3 Activation Increases Vascular Leakage Through Downregulating Tight Junction Proteins: Implications for Diabetic Retinopathy. J. Cell. Physiol. 2017, 232, 1123–1134. [Google Scholar] [CrossRef]
- Amano, W.; Nakajima, S.; Kunugi, H.; Numata, Y.; Kitoh, A.; Egawa, G.; Dainichi, T.; Honda, T.; Otsuka, A.; Kimoto, Y.; et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J. Allergy Clin. Immunol. 2015, 136, 667–677. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/periostin/IL-24 pathway causes epidermal barrier dysfunction in allergic skin inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, P.; Li, F.; Kijlstra, A. The effects of Th17 cytokines on the inflammatory mediator production and barrier function of ARPE-19 cells. PLoS ONE 2011, 6, e18139. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Nusrat, A. Cytokine regulation of tight junctions. Biochim. Biophys. Acta 2009, 1788, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.A.; Thaçi, D.; Hamilton, J.D.; Graham, N.M.; Bieber, T.; Rocklin, R.; Ming, J.E.; Ren, H.; Kao, R.; Simpson, E.; et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N. Engl. J. Med. 2014, 371, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Poumay, Y.; Roland, I.H.; Leclercq-Smekens, M.; Leloup, R. Basal detachment of the epidermis using dispase: Tissue spatial organization and fate of integrin alpha 6 beta 4 and hemidesmosomes. J. Investig. Dermatol. 1994, 102, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Kochkodan, J.J.; Blatt, H.; Lin, S.Y.; Kaplan, N.; Johnston, A.; Swindell, W.R.; Hoover, P.; Schlosser, B.J.; Elder, J.T.; et al. Alteration of the EphA2/Ephrin-A signaling axis in psoriatic epidermis. J. Investig. Dermatol. 2013, 133, 712–722. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brewer, M.G.; Yoshida, T.; Kuo, F.I.; Fridy, S.; Beck, L.A.; De Benedetto, A. Antagonistic Effects of IL-4 on IL-17A-Mediated Enhancement of Epidermal Tight Junction Function. Int. J. Mol. Sci. 2019, 20, 4070. https://doi.org/10.3390/ijms20174070
Brewer MG, Yoshida T, Kuo FI, Fridy S, Beck LA, De Benedetto A. Antagonistic Effects of IL-4 on IL-17A-Mediated Enhancement of Epidermal Tight Junction Function. International Journal of Molecular Sciences. 2019; 20(17):4070. https://doi.org/10.3390/ijms20174070
Chicago/Turabian StyleBrewer, Matthew G., Takeshi Yoshida, Fiona I. Kuo, Sade Fridy, Lisa A. Beck, and Anna De Benedetto. 2019. "Antagonistic Effects of IL-4 on IL-17A-Mediated Enhancement of Epidermal Tight Junction Function" International Journal of Molecular Sciences 20, no. 17: 4070. https://doi.org/10.3390/ijms20174070
APA StyleBrewer, M. G., Yoshida, T., Kuo, F. I., Fridy, S., Beck, L. A., & De Benedetto, A. (2019). Antagonistic Effects of IL-4 on IL-17A-Mediated Enhancement of Epidermal Tight Junction Function. International Journal of Molecular Sciences, 20(17), 4070. https://doi.org/10.3390/ijms20174070