Autism Spectrum Disorder-Related Syndromes: Modeling with Drosophila and Rodents

Abstract
1. Introduction
2. Syndromic Forms of ASD and ASD-Associated Genes
3. Vertebrate Models of ASD-Related Syndromes
3.1. Assay Systems for Rodent ASD Models
3.1.1. Sociability Test
3.1.2. Open Field Behavior Test
3.1.3. Learning and Memory Tests
3.2. Rodent Models of ASD and ASD-Related Syndromes
3.2.1. Non-Genetic Rodent Models of ASD
3.2.2. Genetic Rodent Models of ASD and ASD-Related Syndromes
4. Drosophila Models of ASD and ASD-Related Syndromes
4.1. Assay Systems for Drosophila ASD Models
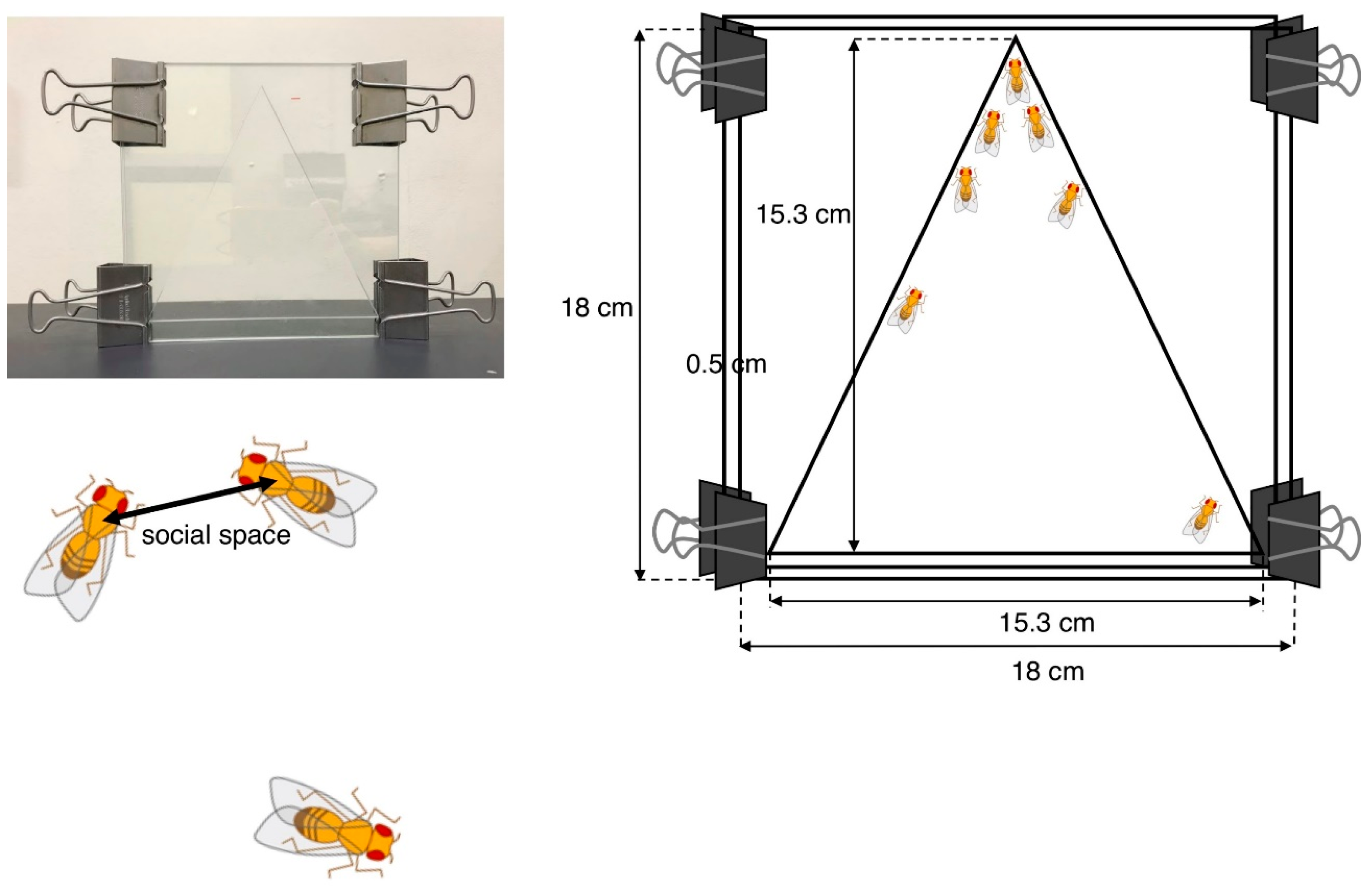
4.1.1. Social Space Assay
4.1.2. Drosophila Activity Assay
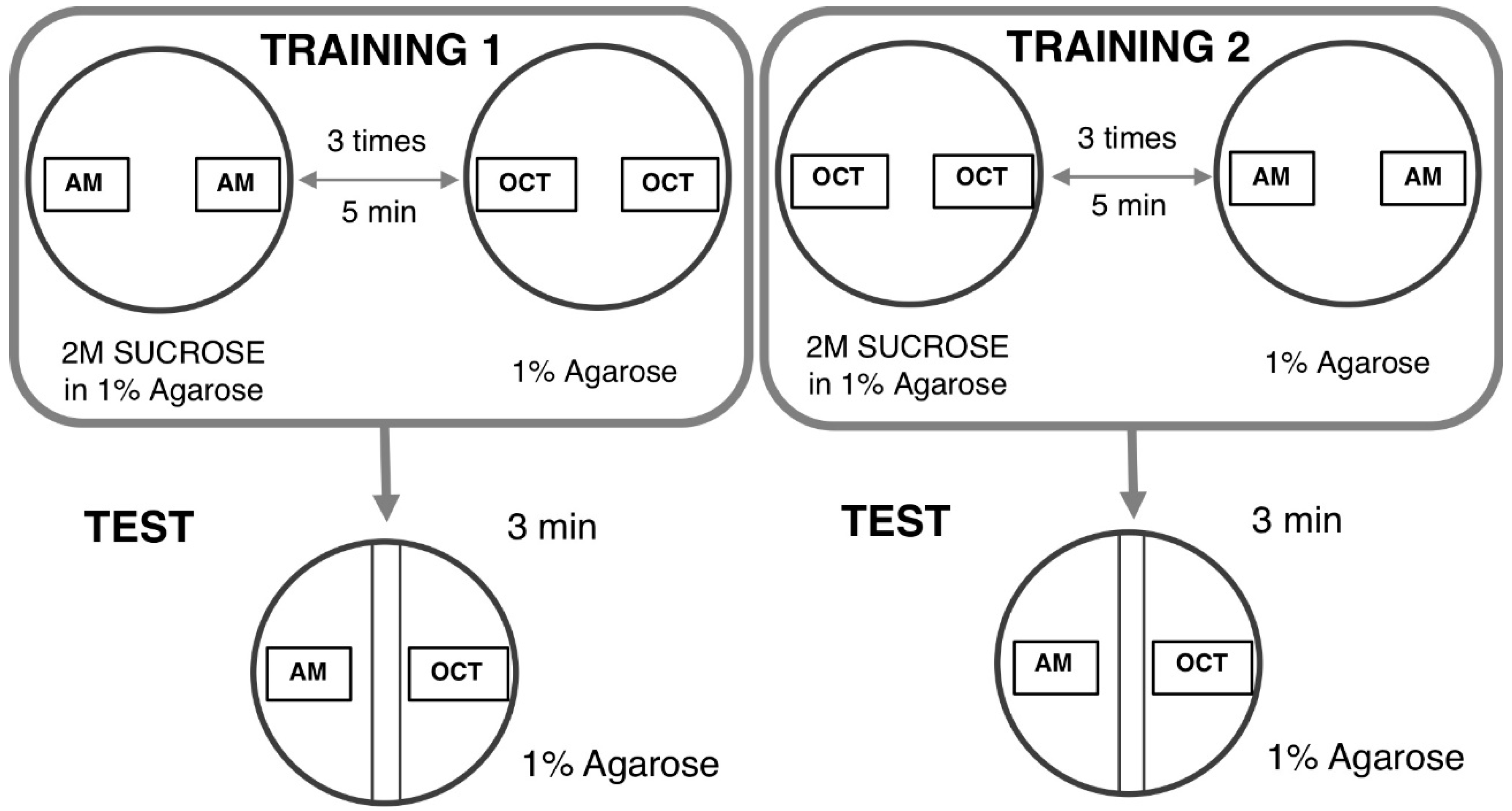
4.1.3. Odor–Taste Learning Assay in Larvae
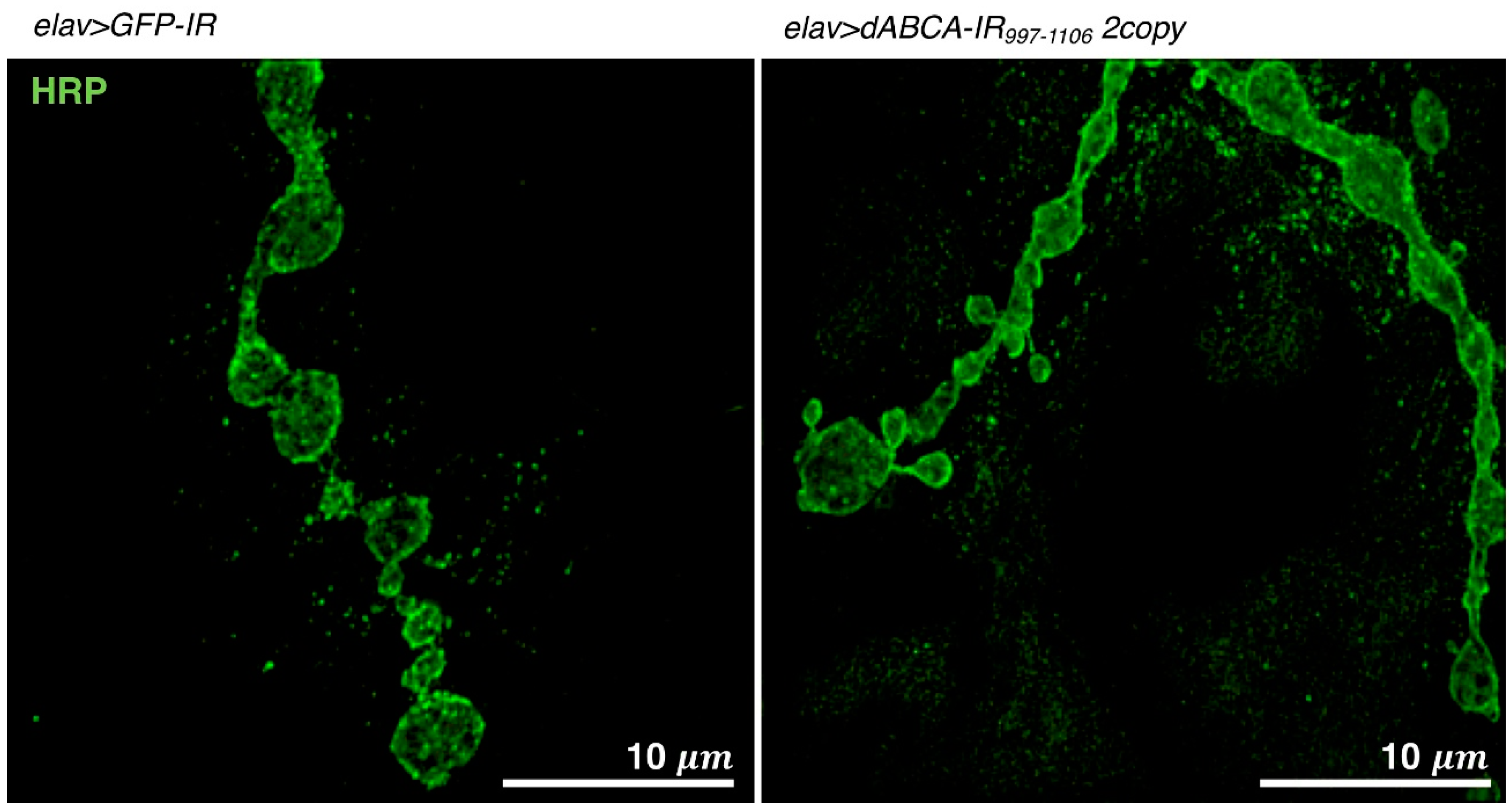
4.1.4. Visualization of NMJs by Super Resolution Microscopy
4.1.5. Electrophysiology at the NMJ
4.2. Drosophila Models Targeting FMR1
4.3. Drosophila Models Targeting UBE3A
4.4. Drosophila Models Targeting Neurobeachin (rugose)
4.5. Drosophila Models Targeting ABCA
4.6. Drosophila ASD and ASD-Related Models Targeting Other Genes
5. Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1418. [Google Scholar] [CrossRef]
- Yuen, R.K.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. Genom. Med. 2016, 1, 160271–1602710. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, Z.C.; Han, J. Drosophila studies on autism spectrum disorders. Nuerosci. Bull. 2017, 33, 737–746. [Google Scholar] [CrossRef]
- Gatto, C.L.; Broadie, K. Drosophila modeling of heritable neurodevelopmental disorders. Curr. Opin. Neurobiol. 2011, 21, 834–841. [Google Scholar] [CrossRef]
- Grice, S.J.; Liu, J.-L.; Webber, C. Synergistic interactions between Drosophila orthologues of genes spanned by de novo human CNVs support multiple-hit models of autism. PLoS Genet. 2015, 11, e1004998. [Google Scholar] [CrossRef]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Dindot, S.V.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008, 17, 111–118. [Google Scholar] [CrossRef]
- Finucane, B.M.; Lusk, L.; Arkilo, D.; Chamberlain, S.; Devinsky, O.; Dindot, S.; Jeste, S.S.; Lasalle, J.M.; Reiter, L.T.; Schanen, N.C.; et al. 15q Duplication Syndrome and Related Disorders; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemia, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; Gene Reviews (R): Seattle, WA, USA, 2016. [Google Scholar]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiple targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2011, 506, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Iwayama, Y.; Maekawa, M.; Toyota, T.; Ohnishi, T.; Toyoshima, M.; Shimamoto, C.; Esaki, K.; Yamada, K.; Iwata, Y.; et al. Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHMT2 and WIZ, in Japanese autism subjects. Mol. Autism 2014, 5, 49. [Google Scholar] [CrossRef]
- Koemans, T.S.; Kleefstra, T.; Chubak, M.C.; Stone, M.H.; Reijnders, M.R.F.; de Munnik, S.; Willemsen, M.H.; Fenckova, M.; Stumpel, C.T.R.M.; Bok, L.A.; et al. Functional convergence of histone methyltransferase EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 2017, 13, e1006864. [Google Scholar] [CrossRef]
- Kilpinen, H.; Ylisaukko-Oja, T.; Hennah, W.; Palo, O.M.; Varilo, T.; Vanhala, R.; Nieminen-von Wendt, T.; von Wendt, L.; Paunio, T.; Peltonen, L. Association of DISC1 with autism and Asperger syndrome. Mol. Psychiatr. 2008, 13, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Fishwick, K.J.; Li, R.A.; Halley, P.; Deng, P.; Storey, K.G. Initiation of neuronal differentiation requires PI3-kinase/TOR signalling in the vertebrate neural tube. Dev. Biol. 2010, 338, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Orlova, K.A.; Crino, P.B. The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 2010, 1184, 87–105. [Google Scholar] [CrossRef]
- Han, S.; Witt, R.M.; Santos, T.M.; Polizzano, C.; Sabatini, B.L.; Ramesh, V. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal. 2008, 20, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Buxbaum, J.D. SHANK3 haploinsufficiency: A ‘‘common’’ but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol. Autism 2013, 4, 17–19. [Google Scholar] [CrossRef]
- Kolevzon, A.; Cai, G.; Soorya, L.; Takahashi, N.; Grodberg, D.; Kajiwara, Y.; Willner, J.P.; Tryfon, A.; Buxbaum, J.D. Analysis of a purported SHANK3 mutation in a boy with autism: Clinical impact of rare variant research in neurodevelopmental disabilities. Brain Res. 2011, 1380, 98–105. [Google Scholar] [CrossRef]
- Arons, M.H.; Thynne, C.J.; Grabrucker, A.M.; Li, D.; Schoen, M.; Cheyne, J.E.; Boeckers, T.M.; Montgomery, J.M.; Garner, C.C. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 2012, 32, 14966–14978. [Google Scholar] [CrossRef]
- Castermans, D.; Wilquet, V.; Parthoens, E.; Huysmans, C.; Steyaert, J.; Swinnen, L.; Fryns, J.P.; Van de Ven, W.; Devriendt, K. The neurobeachin gene is disrupted by a translocation in a patient with idiopathic autism. J. Med. Genet. 2003, 40, 352–356. [Google Scholar] [CrossRef]
- Savelyeva, L.; Sagulenko, E.; Schmitt, J.G.; Schwab, M. The neurobeachin gene spans the common fragile site FRA13A. Hum. Genet. 2006, 118, 551–558. [Google Scholar] [CrossRef]
- Bi, C.; Wu, J.; Jiamg, T.; Liu, Q.; Cai, W.; Yu, P.; Cai, T.; Zhao, M.; Jiang, Y.H.; Sun, Z.S. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum. Mut. 2012, 33, 1635–1638. [Google Scholar] [CrossRef]
- Smith, K.R.; Kopeikina, K.J.; Fawcett-Patel, J.M.; Leaderbrand, K.; Gao, R.; Schurmann, B.; Myczek, K.; Radulovic, J.; Swanson, G.T.; Penzes, P. Psychiatric risk factor ANK3/Ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 2014, 84, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Poot, M. Cennecting the CNTNAP2 networks with neurodevelopmental disorders. Mol. Syndromol. 2015, 6, 7–22. [Google Scholar] [CrossRef]
- Canali, G.; Goutebroze, L. CNTNAP2 heterozygous missense variants: Risk factors for autism spectrum disorder and/or other pathologies? J. Exp. Neurosci. 2018, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sudfof, T.C. Synaptic neurexin complexes: Molecular code for the logic of neural circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Chaste, P.; Leboyer, M. Autism risk factor: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef]
- Nakamura, K.; Anitha, A.; Yamada, K.; Tsujii, M.; Iwayama, Y.; Hattori, E.; Toyota, T.; Suda, S.; Takei, N.; Iwata, Y.; et al. Genetic and expression analyses reveal elevated expression of syntaxin 1A (STX1A) in high functioning autism. Int. J. Neuropsychopharmacol. 2008, 11, 1073–1084. [Google Scholar] [CrossRef]
- Hamilton, P.J.; Campbell, N.G.; Sharma, S.; Erreger, K.; Herborg Hansen, F.; Saunders, C.; Belovich, A.N.; NIH ARRA Autism Sequencing Consortium; Sahai, M.A.; Cook, E.H.; et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 2013, 18, 1315–1323. [Google Scholar] [CrossRef]
- Tomioka, M.; Toda, Y.; Kurisu, J.; Kimura, Y.; Kengaku, M.; Ueda, K. The effects of neurological disorder-related codon variations of ABCA13 on the function of the ABC protein. Biosci. Biotechnol. Biochem. 2012, 76, 2289–2293. [Google Scholar] [CrossRef]
- Knight, H.M.; Pickard, B.S.; Maclean, A.; Malloy, M.P.; Soares, D.C.; McRae, A.F.; Condie, A.; White, A.; Hawkins, W.; McGhee, K.; et al. A cytogenetic abnormality and rare coding variants identify ABCA13 as a candidate gene in schizophrenia, bipolar disorder, and depression. Am. J. Hum. Genet. 2009, 85, 833–846. [Google Scholar] [CrossRef]
- Yoshida, K.; Go, Y.; Kushima, I.; Toyoda, A.; Fujiyama, A.; Imai, H.; Saito, N.; Iriki, A.; Ozaki, N.; Isoda, M. Single-neuron and genetic correlates of autistic behavior in macaque. Sci. Adv. 2016, 2, e1600558. [Google Scholar] [CrossRef]
- Jobski, K.; Hofer, J.; Hoffmann, F.; Bachmann, C. Use of psychotropic drugs in patients with autism spectrum disorders: A systematic review. Acta Psychiatr. Scand. 2017, 135, 8–28. [Google Scholar] [CrossRef]
- Lord, C.; Risi, S.; Lambrecht, L.; Cook, E.H., Jr.; Leventhal, B.L.; DiLavore, P.C.; Pickles, A.; Rutter, M. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, F.R.; Pauls, D. Autism. Lancet 2003, 362, 1133–1141. [Google Scholar] [CrossRef]
- Okada, R.; Fujiwara, H.; Mizuki, D.; Araki, R.; Yabe, T.; Matsumoto, K. Involvement of dopaminergic and cholinergic systems in social isolation-induced deficits in social affiliation and conditional fear memory in mice. Neuroscience 2015, 299, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, K.; Fujiwara, H.; Awale, S.; Dibwe, D.F.; Araki, R.; Yabe, T.; Matsumoto, K. Decrease in endogenous brain allopregnanolone induces autism spectrum disorder (ASD)-like behavior in mice: A novel animal model of ASD. Behav. Brain Res. 2017, 334, 6–15. [Google Scholar] [CrossRef]
- Fujiwara, H.; Han, Y.; Ebihara, K.; Awale, S.; Araki, R.; Yabe, T.; Matsumoto, K. Daily administration of yokukansan and keishito prevents social isolation-induced behavioral abnormalities and down-regulation of phosphorylation of neuroplasticity-related signaling molecules in mice. BMC Complement. Altern. Med. 2017, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mousemodels of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, H.; Ono, K.; Murakami, Y.; Matsumoto, K. Social isolation induces deficit of latent learning performance in mice: A putative animal model of attention deficit/hyperactivity disorder. Behav. Brain Res. 2013, 238, 146–153. [Google Scholar] [CrossRef]
- Riedel, G.; Spink, A.; Veltkamp, R. Measuring Behavior. J. Neurosci. Methods 2014, 234, 127–134. [Google Scholar]
- Mamiya, T.; Noda, Y.; Nishi, M.; Takeshima, H.; Nabeshima, T. Enhancement of spatial attention in nociceptin/orphanin FQ receptor-knockout mice. Brain Res. 1998, 783, 236–240. [Google Scholar] [CrossRef]
- Gepner, B.; Feron, F. Autism: A world changing too fast for a mis-wired brain? Neurosci. Biobehav. Rev. 2009, 33, 1227–1242. [Google Scholar] [CrossRef]
- Roullet, F.I.; Lai, J.K.; Foster, J.A. In utero exposure to valproic acid and autism-a current review of clinical and animal studies. Neurotoxicol. Teratol. 2013, 36, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Al-Houqani, M.; Sadeq, A.; Ojha, S.K.; Sasse, A.; Sadek, B. Current enlightenment about etiology and pharmacological treatment of autism spectrum disorder. Front. Neurosci. 2018, 12, 304. [Google Scholar] [CrossRef]
- Bolte, S.; Girdler, S.; Marschik, P.B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell. Mol. Life Sci. 2019, 76, 1275–1297. [Google Scholar] [PubMed]
- Christianson, A.L.; Chesler, N.; Kromberg, J.G.R. Fetal valproate syndrome: Clinical and neurodevelopmental features in two sibling pairs. Dev. Med. Child Neurol. 1994, 36, 357–369. [Google Scholar]
- Strömland, K.; Nordin, V.; Miller, M.T.; Akerstrom, B.; Gillberg, C. Autism in thalidomide embryopathy: A population study. Dev. Med. Child Neurol. 1994, 36, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Nanson, J.L. Autism in fetal alcohol syndrome: A report of six cases. Alcohol. Clin. Exp. Res. 1992, 16, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Turnpenny, P.; Quinn, A.; Glover, S.; Lloyd, D.J.; Montgomery, T.; Dean, J.C. A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 2000, 37, 489–497. [Google Scholar] [CrossRef]
- Meador, K.J.; Baker, G.A.; Browning, N.; Cohen, M.J.; Clayton-Smith, J.; Kalayjian, L.A.; Kanner, A.; Liporace, J.D.; Pennell, P.B.; Privitera, M.; et al. NEAD Study Group. Foetal antiepileptic drug exposure and verbal versus non-verbal abilities at three years of age. Brain 2011, 134, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Shallcross, R.; Bromley, R.L.; Irwin, B.; Bonnett, L.J.; Morrow, J.; Baker, G.A. Child development following in utero exposure: Levetiracetam vs sodium valproate. Neurology 2011, 76, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Bambini-Junior, V.; Rodrigues, L.; Behr, G.A.; Moreira, J.C.; Riesgo, R.; Gottfried, C. Animal model of autism induced by prenatal exposure to valproate: Behavioral changes and liver parameters. Brain Res. 2011, 1408, 8–16. [Google Scholar] [CrossRef]
- Massa, V.; Cabrera, R.M.; Menegola, E.; Giavini, E.; Finnell, R.H. Valproic acid-induced skeletal malformations: Associated gene expression cascades. Pharm. Genom. 2005, 15, 787–800. [Google Scholar] [CrossRef]
- Kogan, M.D.; Vladutiu, C.J.; Schieve, L.A.; Ghandour, R.M.; Blumberg, S.J.; Zablotsky, B.; Perrin, J.M.; Shattuck, P.; Kuhlthau, K.A.; Harwood, R.L.; et al. The prevalence of parent-reported autism spectrum disorder among US children. Pediatrics 2018, 142, e20174161. [Google Scholar] [CrossRef]
- Kim, K.C.; Kim, P.; Go, H.S.; Choi, C.S.; Park, J.H.; Kim, H.J.; Jeon, S.J.; Dela Pena, I.C.; Han, S.H.; Cheong, J.H.; et al. Male-specific alteration in excitatory post-synaptic development and social interaction in pre-natal valproic acid exposure model of autism spectrum disorder. J. Neurochem. 2013, 124, 832–843. [Google Scholar] [CrossRef]
- Kataoka, S.; Takuma, K.; Hara, Y.; Maeda, Y.; Ago, Y.; Matsuda, T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int. J. Neuropsychopharmacol. 2013, 16, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Roman, A.; Basta-Kaim, A.; Kubera, M.; Budziszewska, B.; Schneider, K.; Przewłocki, R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology 2008, 33, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Gonzales, E.L.; Mabunga, D.F.N.; Valencia, S.T.; Kim, D.G.; Kim, Y.; Adil, K.J.L.; Shin, D.; Park, D.; Shin, C.Y. Sex-specific Behavioral Features of Rodent Models of Autism Spectrum Disorder. Exp. Neurobiol. 2018, 27, 321–343. [Google Scholar] [CrossRef]
- Kuo, H.Y.; Liu, F.C. Molecular pathology and pharmacological treatment of autism spectrum disorder-like phenotypes using rodent models. Front. Cell Neurosci. 2018, 12, 422. [Google Scholar] [CrossRef]
- Puia, G.; Mienville, J.M.; Matsumoto, K.; Takahata, H.; Watanabe, H.; Costa, E.; Guidotti, A. On the putative physiological role of allopregnanolone on GABAA receptor function. Neuropharmacology 2003, 44, 49–55. [Google Scholar] [CrossRef]
- Coghlan, S.; Horder, J.; Inkster, B.; Mendez, M.A.; Murphy, D.G.; Nutt, D.J. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci. Biobehav. Rev. 2012, 36, 2044–2055. [Google Scholar] [CrossRef]
- Cellot, G.; Cherubini, E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2014, 2, 70. [Google Scholar] [CrossRef]
- Guo, Q.-Y.; Ebihara, K.; Shimodaira, T.; Fujiwara, H.; Toume, K.; Dibwe, D.F.; Awale, S.; Araki, R.; Yabe, T.; Matsumoto, K. Kami-shoyo-san improves ASD-like behaviors caused by decreasing allopregnanolone biosynthesis in an SKF mouse model of autism. PLoS ONE 2019, 14, e0211266. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, Y.; Sadakata, T.; Furuichi, T. Animal models of autism spectrum disorder (ASD): A synaptic-level approach to autistic-like behavior in mice. Exp. Anim. 2013, 62, 71–78. [Google Scholar] [CrossRef] [PubMed]
- de la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [PubMed]
- Hubert, S.W.; Jiang, Y.H. Monogenic mouse models of autism spectrum disorders: Common mechanisms and missing links. Neuroscience 2016, 321, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Dahlhaus, R.; El-Husseini, A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 2010, 208, 96–105. [Google Scholar] [CrossRef]
- Spencer, C.M.; Alekseyenko, O.; Hamilton, S.M.; Thomas, A.M.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Modifying behavioral phenotypes in Fmr1 KO mice: Genetic background difference reveal autistic-like responses. Autism Res. 2011, 4, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Samaco, R.C.; McGraw, C.M.; Ward, C.S.; Sun, Y.; Neul, J.L.; Zoghbi, H.Y. Female Mecp2+/− mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 2012, 22, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Itoh, M.; Ichikawa, T.; Washiyama, K.; Goto, Y.J. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. Neuropathol. Exp. Neurol. 2005, 64, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Kline, D.D.; Ogier, M.; Kunze, D.L.; Katz, D.M. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J. Neurosci. 2010, 30, 5303–5310. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef]
- Miao, S.; Chen, R.; Ye, J.; Tan, G.H.; Li, S.; Zhang, J.; Jiang, Y.H.; Xiang, Z.Q. The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci. 2013, 33, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef]
- Cao, C.; Rioult-Pedotti, M.S.; Migani, P.; Yu, C.J.; Tiwari, R.; Parang, K.; Spaller, M.R.; Goebel, D.J.; Marshall, J. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 2013, 11, e1001478. [Google Scholar] [CrossRef]
- Bey, A.L.; Jiang, Y.H. Overview of mouse models of autism spectrum disorders. Curr. Protoc. Pharmacol. 2014, 66, 1–26. [Google Scholar]
- Crino, P.B. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013, 125, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Nuytens, K.; Gantois, I.; Stijnen, P.; Iscru, E.; Laeremans, A.; Serneels, L.; Van Eylen, L.; Liebhaber, S.A.; Devriendt, K.; Balschun, D.; et al. Haploinsufficiency of the autism candidate gene Neurobeachin induces autism-like behaviors and affects cellular and molecular processes of synaptic plasticity in mice. Neurobiol. Dis. 2013, 51, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Niesmann, K.; Breuer, D.; Brockhaus, J.; Born, G.; Wolff, I.; Reissner, C.; Kilimann, M.W.; Rohlmann, A.; Missler, M. Dendritic spine formation and synaptic function require neurobeachin. Nat. Commun. 2011, 2, 557. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.; Lauks, J.; Jung, S.; Cooke, N.E.; de Wit, H.; Brose, N.; Kiliman, M.W.; Verhage, M.; Rhee, J. Neurobeachin regulates neurotransmitter receptor trafficking to synapse. J. Cell Biol. 2013, 200, 62–80. [Google Scholar] [CrossRef]
- Volders, K.; Nuytens, K.; Creemers, J.W. The autism candidate gene Neurobeachin encodes a scaffolding protein implicated in membrane trafficking and signaling. Curr. Mol. Med. 2011, 11, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.F.; Chou, M.T.; Salazar, E.D.; Nicholson, T.; Saini, N.; Metchev, S.; Krantz, D.E. A simple assay to study social behavior in Drosophila: Measurement of social space within a group. Genes Brain Behav. 2012, 11, 243–252. [Google Scholar] [CrossRef]
- Hamilton, P.J.; Campbell, N.G.; Sharma, S.; Erreger, K.; Hansen, F.H.; Saunders, C.; Belovich, A.N.; Sahai, M.A.; Cook, E.H.; Gether, U.; et al. Drosophila melanogaster: A novel animal model for the behavioral characterization of autism-associated mutations in the dopamine transporter gene. Mol. Psychiatry 2013, 18, 1235. [Google Scholar] [CrossRef] [PubMed]
- Ueoka, I.; Kawashima, H.; Konishi, A.; Aoki, M.; Tanaka, R.; Yoshida, H.; Maeda, T.; Ozaki, M.; Yamaguchi, M. Novel Drosophila model for psychiatric disorders including autism spectrum disorder by targeting of ATP-binding cassette protein A. Exp. Neurol. 2018, 300, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Gerber, B.; Biernacki, R.; Thum, J. Oor-taste learning assays in Drosophila larvae. Cold Spring Harb. Protoc. 2013, 8, 213–223. [Google Scholar]
- Jantrapirom, S.; Lo Piccolo, L.; Yoshida, H.; Yamaguchi, M. A new Drosophila model of Ubiquilin knockdown shows the effect of impaired proteostasis on locomotive and learning abilities. Exp. Cell. Res. 2018, 362, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Takashima, H. Drosophila Charcot-Marie-Tooth disease models. Adv. Exp. Med. Biol. 2018, 1076, 97–117. [Google Scholar] [PubMed]
- Brent, J.R.; Werner, K.M.; McCabe, B.D. Drosophila larval NMJ dissection. J. Vis. Exp. 2009, 24. [Google Scholar] [CrossRef] [PubMed]
- Potikanond, S.; Nimlamool, W.; Noordermeer, J.; Fradkin, L.G. Muscular Dystrophy Model. Adv. Exp. Med. Biol. 2018, 1076, 147–172. [Google Scholar]
- Wan, L.; Dockendorff, T.C.; Jongens, T.A.; Dreyfuss, G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell Biol. 2000, 20, 8536–8547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Bailey, A.M.; Matthies, H.J.; Renden, R.B.; Smith, M.A.; Speese, S.D.; Rubin, G.M.; Broadie, K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 2001, 107, 591–603. [Google Scholar] [CrossRef]
- Morales, J.; Hiesinger, P.R.; Schroeder, A.J.; Kume, K.; Verstreken, P.; Jackson, F.R.; Nelson, D.L.; Hassan, B.A. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 2002, 34, 961–972. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, Y.Q.; Woodruff, E.; Broadie, K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 2004, 14, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Coffee, R.L.; Tessier, C.R.; Woodruff, E.A.; Broadie, K. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis. Model. Mech. 2010, 3, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Gatto, C.L.; Broadie, K. Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 2008, 135, 2637–2648. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Li, W.; Xu, K.; Bogert, B.A.; Su, K.; Gao, F.B. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 2003, 130, 5543–5552. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Li, Y.; Wang, F.; Gao, F.B. The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J. Neurosci. 2008, 28, 11883–11889. [Google Scholar] [CrossRef]
- Dockendorff, T.C.; Su, H.S.; McBride, S.M.; Yang, Z.; Choi, C.H.; Siwicki, K.K.; Sehgal, A.; Jongens, T.A. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 2002, 34, 973–984. [Google Scholar] [CrossRef]
- Tessier, C.R.; Broadie, K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 2008, 135, 1547–1557. [Google Scholar] [CrossRef]
- Michel, C.I.; Kraft, R.; Restifo, L.L. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J. Neurosci. 2004, 24, 5798–5809. [Google Scholar] [CrossRef]
- Kashima, R.; Roy, S.; Ascano, M.; Martinez-Cerdeno, V.; Ariza-Torres, J.; Kim, S.; Louie, J.; Lu, Y.; Leyton, P.; Bloch, K.D.; et al. Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci. Signal. 2016, 9, ra58. [Google Scholar] [CrossRef]
- Cvetkovska, V.; Hibbert, A.D.; Emran, F.; Chen, B.E. Overexpression of Down syndrome cell adhesion molecule impairs precise synaptic targeting. Nat. Neurosci. 2013, 16, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Sudhakaran, I.P.; Hillebrand, J.; Dervan, A.; Das, S.; Holohan, E.E.; Hülsmeier, J.; Sarov, M.; Parker, R.; VijayRaghavan, K.; Ramaswami, M. FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proc. Natl. Acad. Sci. USA 2014, 111, E99–E108. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Jepson, J.E.; Savva, Y.A.; Pepper, A.S.; Reenan, R.A.; Jongens, T.A. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat. Neurosci. 2011, 14, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, I.; Rodríguez-Revenga, L.; Xunclà, M.; Milà, M. 15q11.2 microdeletion and FMR1 premutation in a family with intellectual disabilities and autism. Gene 2012, 508, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Waltes, R.; Duketis, E.; Knapp, M.; Anney, R.J.L.; Huguet, G.; Schlitt, S.; Jarczok, T.A.; Sachse, M.; Kämpfer, L.M.; Kleinböck, T.; et al. Common variants in genes of the postsynaptic FMRP signaling pathway are risk factors for autism spectrum disorders. Hum. Genet. 2014, 133, 781–792. [Google Scholar] [CrossRef]
- Huang, Y. Up-regulated cytoplasmic FMRP-interacting protein 1 in intractable temporal lobe epilepsy patients and a rat model. Int. J. Neurosci. 2015, 126, 542–551. [Google Scholar] [CrossRef]
- Wang, J.; Tao, Y.; Song, F.; Sun, Y.; Ott, J.; Saffen, D. Common regulatory variants of CYFIP1 contribute to susceptibility for Autism Spectrum Disorder (ASD) and classical autism. Ann. Hum. Genet. 2015, 79, 329–340. [Google Scholar] [CrossRef]
- Abekhoukh, S.; Sahin, H.B.; Grossi, M.; Zongaro, S.; Maurin, T.; Madrigal, I.; Kazue-Sugioka, D.; Raas-Rothschild, A.; Doulazmi, M.; Carrera, P.; et al. New insights into the regulatory function of CYFIP1 in the context of WAVE- and FMRP-containing complexes. Dis. Model. Mech. 2017, 10, 463–474. [Google Scholar] [CrossRef]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling fragile X syndrome in Drosophila. Front. Mol. Neurosci. 2018, 11, 124. [Google Scholar] [CrossRef]
- Johnson, K.G.; Tenney, A.P.; Ghose, A.; Duckworth, A.M.; Higashi, M.E.; Parfitt, K.; Marcu, O.; Heslip, T.R.; Marsh, J.L.; Schwarz, T.L.; et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron 2006, 49, 517–531. [Google Scholar] [CrossRef]
- Friedman, S.H.; Dani, N.; Rushton, E.; Broadie, K. Fragile X mental retardation protein regulates trans-synaptic signaling in Drosophila. Dis. Model Mech. 2013, 6, 1400–1413. [Google Scholar] [CrossRef]
- Choi, C.H.; McBride, S.M.J.; Schoenfeld, B.P.; Liebelt, D.A.; Ferreiro, D.; Ferrick, N.J.; Hinchey, P.; Kollaros, M.; Rudominer, R.L.; Terlizzi, A.M.; et al. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 2010, 11, 347–362. [Google Scholar] [CrossRef]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive locomotion in a Drosophila model is a functional readout for the synaptic abnormalities underlying fragile X syndrome. Sci. Signal. 2017, 10, eaai8133. [Google Scholar] [CrossRef] [PubMed]
- Monyak, R.E.; Emerson, D.; Schoenfeld, B.P.; Zheng, X.; Chambers, D.B.; Rosenfelt, C.; Langer, S.; Hinchey, P.; Choi, C.H.; McDonald, T.V.; et al. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 2016, 22, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Seagroves, T.N.; Bowers, M.; Bier, E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum. Mol. Genet. 2006, 15, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Bolduc, F.V.; Bell, K.; Tully, T.; Fang, Y.; Sehgal, A.; Fischer, J.A. A Drosophila model for Angelman syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 12399–12404. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, F.; Li, Y.; Ferris, J.; Lee, J.A.; Gao, F.B. The Drosophila homologue of the Angelman syndrome ubiquitin ligase regulates the formation of terminal dendritic branches. Hum. Mol. Genet. 2009, 18, 454–462. [Google Scholar] [CrossRef]
- Chakraborty, M.; Paul, B.K.; Nayak, T.; Das, A.; Jana, N.R.; Bhutani, S. The E3 ligase ube3a is required for learning in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2015, 462, 71–77. [Google Scholar] [CrossRef]
- Hope, K.A.; LeDoux, M.S.; Reiter, L.T. Glial overexpression of Dube3a causes seizures and synaptic impairments in Drosophila concomitant with down regulation of the Na+/K+ pump ATPα. Neurobiol. Dis. 2017, 108, 238–248. [Google Scholar] [CrossRef]
- Valdez, C.; Scroggs, R.; Chassen, R.; Reiter, L.T. Variation in Dube3a expression affects neurotransmission at the Drosophila neuromuscular junction. Biol. Open 2015, 4, 776–782. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferdousy, F.; Bodeen, W.; Summers, K.; Doherty, O.; Wright, O.; Elsisi, N.; Hilliard, G.; O’Donnell, J.M.; Reiter, L.T. Drosophila Ube3a regulates monoamine synthesis by increasing GTP cyclohydrolase I activity via a non-ubiquitin ligase mechanism. Neurobiol. Dis. 2011, 41, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Bucan, M.; Abrahams, B.S.; Wang, K.; Glessner, J.T.; Herman, E.I.; Sonnenblick, L.I.; Alvarez Retuerto, A.I.; Imielinski, M.; Hadley, D.; Bradfield, J.P.; et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009, 5, e1000536. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ramirez, J.; Franco, M.; Lectez, B.; Gonzalez, M.; Barrio, R.; Mayor, U. Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life. Sci. 2014, 71, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Tuand, K.; Stijnen, P.; Volders, K.; Declercq, J.; Nuytens, K.; Meulemans, S.; Creemers, J. Nuclear localization of the autism candidate gene neurobeachin and functional interaction with the NOTCH1 intracellular domain indicate a role in regulating transcription. PLoS ONE 2016, 11, e0151954. [Google Scholar] [CrossRef] [PubMed]
- Volders, K.; Scholz, S.; Slabbaert, J.R.; Nagel, A.C.; Verstreken, P.; Creemers, J.W.M.; Callaerts, P.; Schwarzel, M. Drosophila rugose is a functional homolog of mammalian neurobeachin and affects synaptic architecture, brain morphology, and associative learning. J. Neurosci. 2012, 32, 15193–15204. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.; Tenezaca, L.; Fernandez, R.W.; Schatoff, E.; Flores, J.; Ueda, A.; Zhong, X.; Wu, C.-F.; Simon, A.F.; Venkatesh, T. Drosophila mutants of the autism candidate gene neurobeachin (rugose) exhibit neuro-developmental disorders, aberrant synaptic properties, altered locomotion, impaired adult social behavior and activity patterns. J. Neurogenet. 2015, 29, 135–143. [Google Scholar] [CrossRef]
- Shamloula, H.K.; Mbogho, M.P.; Pimentel, A.C.; Chrzanowska-Lightowlers, Z.M.A.; Hyatt, V.; Okano, H.; Venkatesh, T.R. Rugose (rg), a Drosophila A kinase anchor protein, is required for retinal pattern formation and interacts genetically with multiple signaling pathways. Genetics 2002, 161, 693–710. [Google Scholar]
- Wech, I.; Nagel, A.C. Mutations in rugose promote cell type-specific apoptosis in the Drosophila eye. Cell Death Differ. 2005, 12, 145–152. [Google Scholar] [CrossRef]
- Iritani, S.; Torii, Y.; Habuchi, C.; Sekiguchi, H.; Fujishiro, H.; Yoshida, M.; Go, Y.; Iriki, A.; Isoda, M.; Ozaki, N. The neuropathological investigation of the brain in a monkey model of autism spectrum disorder with ABCA13 deletion. Int. J. Dev. Neurosci. 2018, 71, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Piehler, A.P.; Özcurumez, M.; Kaminski, W.E. A-subclass ATP-binding cassette proteins in brain lipid homeostasis and neurodegeneration. Front. Psychiatry 2012, 3, 1–16. [Google Scholar] [CrossRef]
- Seeman, P.; Niznik, H.B. Dopamine receptors and transporters in Parkinson’s disease and schizophrenia. FASEB J. 1990, 4, 2737–2744. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Newcorn, J.; Telang, F.; Solanto, M.V.; Fowler, J.S.; Logan, J.; Ma, Y.; Schulz, K.; Pradhan, K.; et al. Depressed dopamine activity in caudate and preliminary evidence of limbic involvement in adults with attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 2007, 64, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Gadow, K.D.; Roohi, J.; DeVincent, C.J.; Hatchwell, E. Association of ADHD, tics, and anxiety with dopamine transporter (DAT1) genotype in autism spectrum disorder. J. Child Psychol. Psychiatry 2008, 49, 1331–1338. [Google Scholar] [CrossRef]
- Cousins, D.A.; Butts, K.; Young, A.H. The role of dopamine in bipolar disorder. Bipolar Disord. 2009, 11, 787–806. [Google Scholar] [PubMed]
- Nakamura, K.; Sekine, Y.; Ouchi, Y.; Tsujii, M.; Yoshikawa, E.; Futatsubashi, M.; Tsuchiya, K.J.; Sugihara, G.; Iwata, Y.; Suzuki, K.; et al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch. Gen. Psychiatry 2010, 67, 59–68. [Google Scholar] [CrossRef]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef]
- Kumar, R.A.; KaraMohamed, S.; Sudi, J.; Conrad, D.F.; Brune, C.; Badner, J.A.; Gilliam, T.C.; Nowak, N.J.; Cook, E.H., Jr.; Dobyns, W.B.; et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008, 17, 628–638. [Google Scholar] [CrossRef]
- Horev, G.; Ellegood, J.; Lerch, J.P.; Son, Y.E.; Muthuswamy, L.; Vogel, H.; Krieger, A.M.; Buja, A.; Henkelman, R.M.; Wigler, M.; et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA 2011, 108, 17076–17081. [Google Scholar] [CrossRef]
- Park, S.M.; Littleton, J.T.; Park, H.R.; Lee, J.H. Drosophila homolog of human KIF22 at the autism-linked 16p11.2 loci influences synaptic connectivity at larval neuromuscular junctions. Exp. Neurobiol. 2016, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Park, H.R.; Lee, J.H. MAPK3 at the autism-linked human 15p11.2 locus influences precise synaptic target selection at Drosophila larval neuromuscular junctions. Mol. Cells 2017, 40, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kusenda, M.; Sebat, J. The role of rare structural variants in the genetics of autism spectrum disorders. Cytogenet. Genome Res. 2008, 123, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Moreno-De-Luca, D.; Sanders, S.J.; Willsey, A.J.; Mulle, J.G.; Lowe, J.K.; Geschwind, D.H.; State, M.W.; Martin, C.L.; Ledbetter, D.H. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 2013, 18, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.J.; Ponting, C.P.; Boulding, H.C.; Meader, S.; Betancur, C.; Buxbaum, J.D.; Pinto, D.; Marshall, C.R.; Lionel, A.C.; Scherer, S.W.; et al. Network topologies and convergent aetiologies arising from deletions and duplications observed in individuals with autism. PLoS Genet. 2013, 9, e1003523. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse | Drosophila | |
|---|---|---|
| Genome size | 2.8 Gbp | 0.14 Gbp |
| Protein coding genes shared with human | 88% | 58% |
| % of human disease genes conserved | 99% | 75% |
| Neuronal cells/brain | 7 × 107 | 1.35 × 105 |
| Complex behavior | +++ | + |
| Generation time | 50 days | 10 days |
| Genome wide genetic screen | + | +++ |
| Production of offspring/female | 10/litter | 100 eggs/day |
| Ethical restriction | +++ | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueoka, I.; Pham, H.T.N.; Matsumoto, K.; Yamaguchi, M. Autism Spectrum Disorder-Related Syndromes: Modeling with Drosophila and Rodents. Int. J. Mol. Sci. 2019, 20, 4071. https://doi.org/10.3390/ijms20174071
Ueoka I, Pham HTN, Matsumoto K, Yamaguchi M. Autism Spectrum Disorder-Related Syndromes: Modeling with Drosophila and Rodents. International Journal of Molecular Sciences. 2019; 20(17):4071. https://doi.org/10.3390/ijms20174071
Chicago/Turabian StyleUeoka, Ibuki, Hang Thi Nguyet Pham, Kinzo Matsumoto, and Masamitsu Yamaguchi. 2019. "Autism Spectrum Disorder-Related Syndromes: Modeling with Drosophila and Rodents" International Journal of Molecular Sciences 20, no. 17: 4071. https://doi.org/10.3390/ijms20174071
APA StyleUeoka, I., Pham, H. T. N., Matsumoto, K., & Yamaguchi, M. (2019). Autism Spectrum Disorder-Related Syndromes: Modeling with Drosophila and Rodents. International Journal of Molecular Sciences, 20(17), 4071. https://doi.org/10.3390/ijms20174071