A SNP Mutation of SiCRC Regulates Seed Number Per Capsule and Capsule Length of cs1 Mutant in Sesame


Abstract
:1. Introduction
2. Results
2.1. Phenotypic Comparison
2.2. Inheritance Analysis
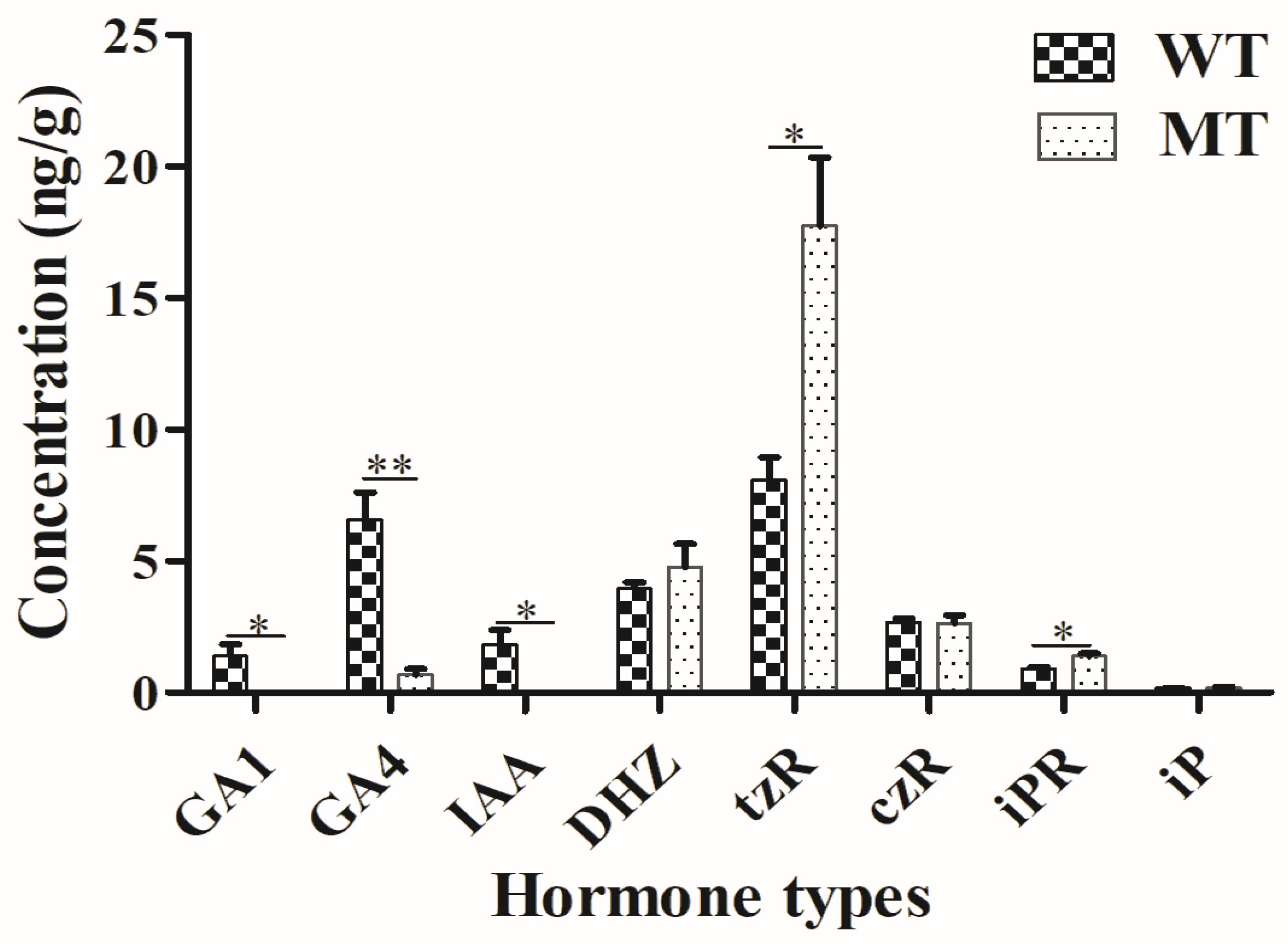
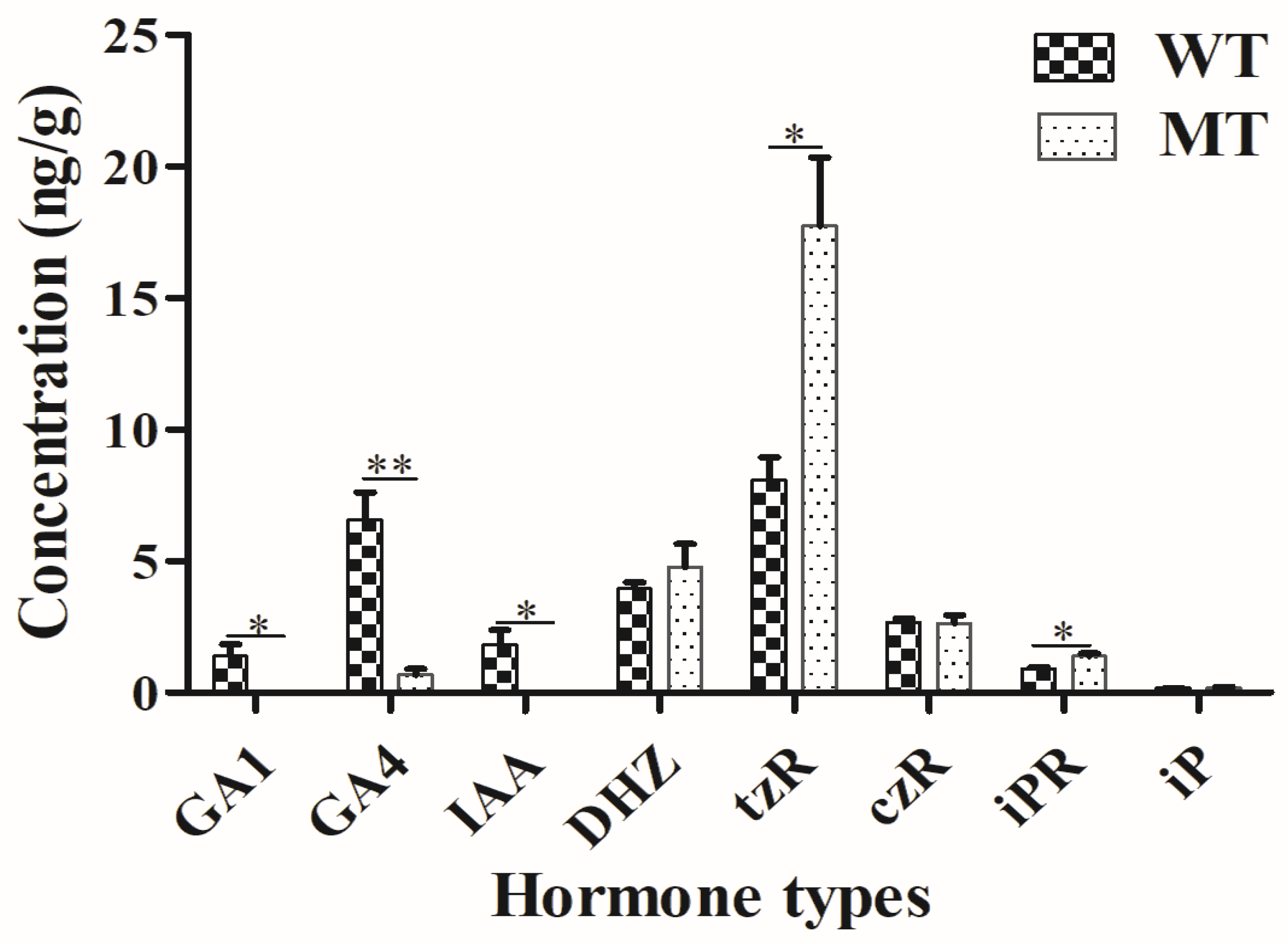
2.3. Plant Hormone Determination
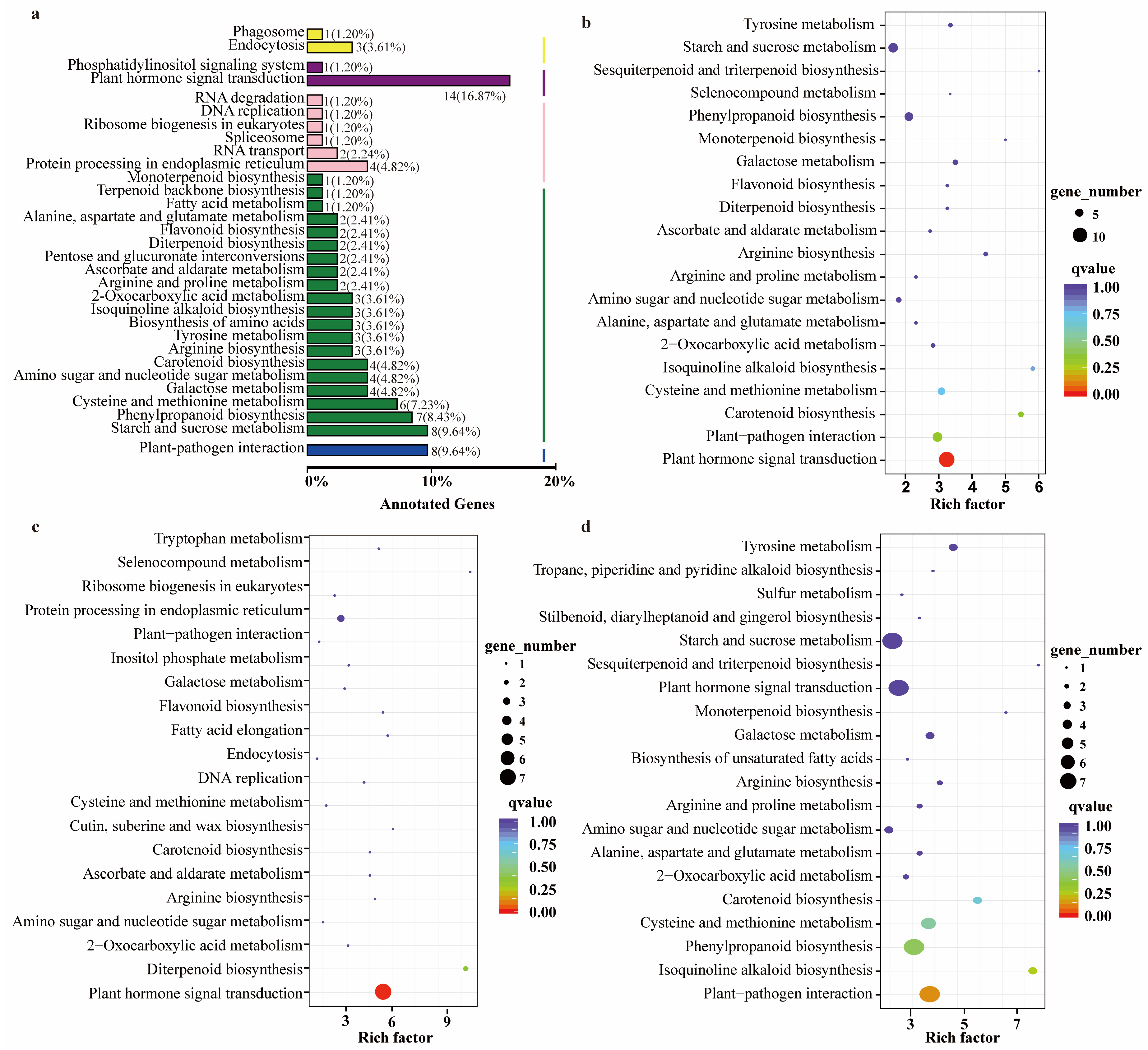
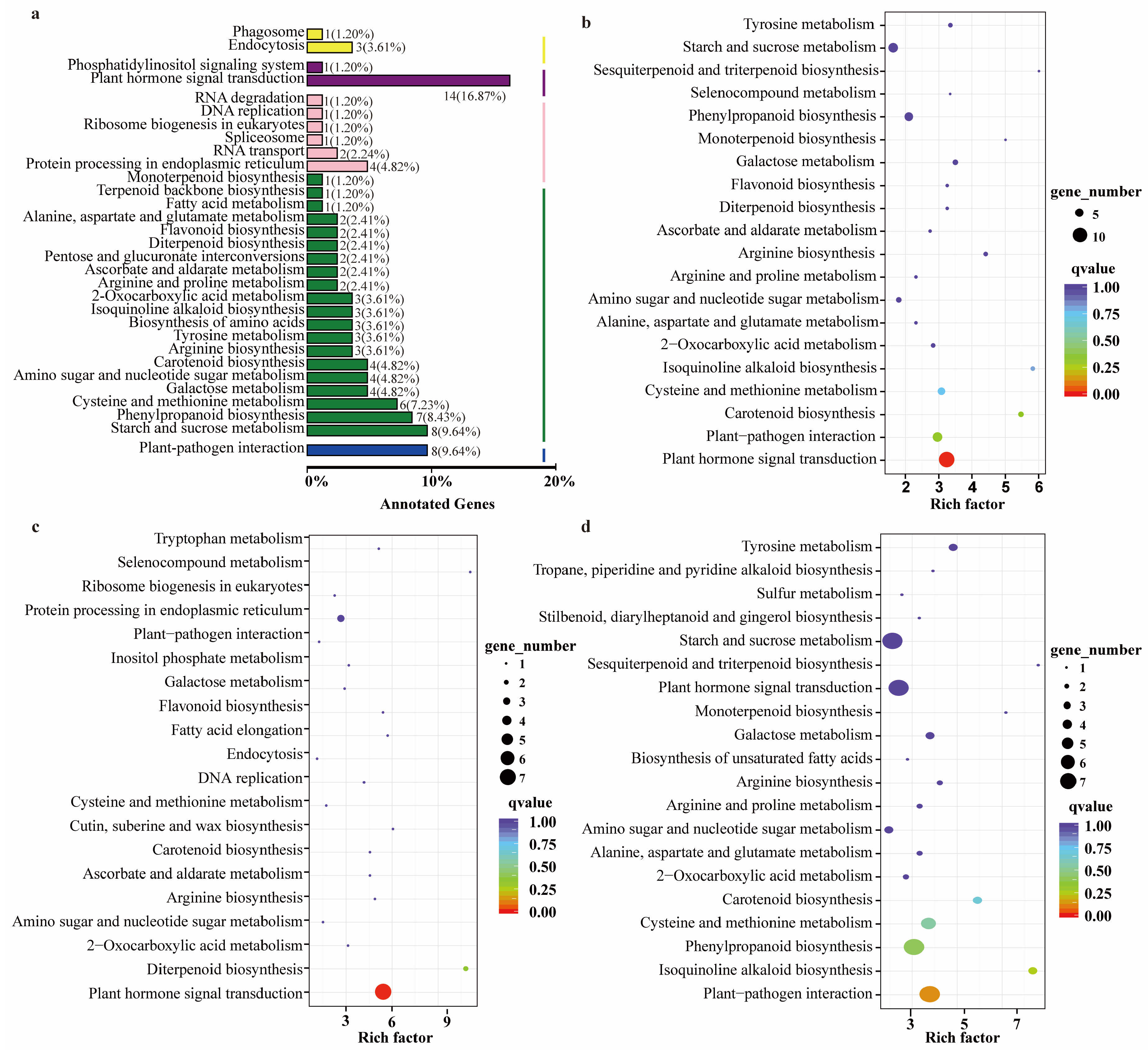
2.4. RNA-Seq Analysis
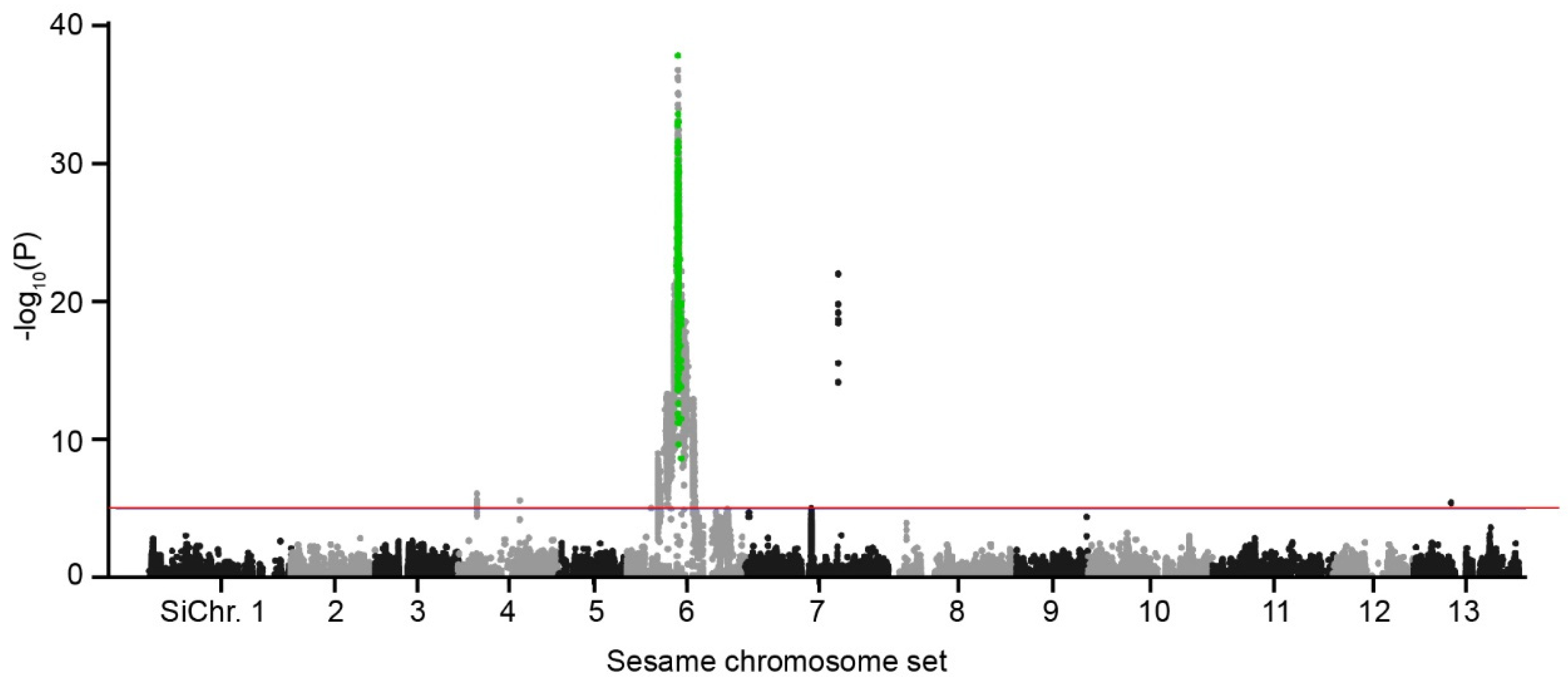
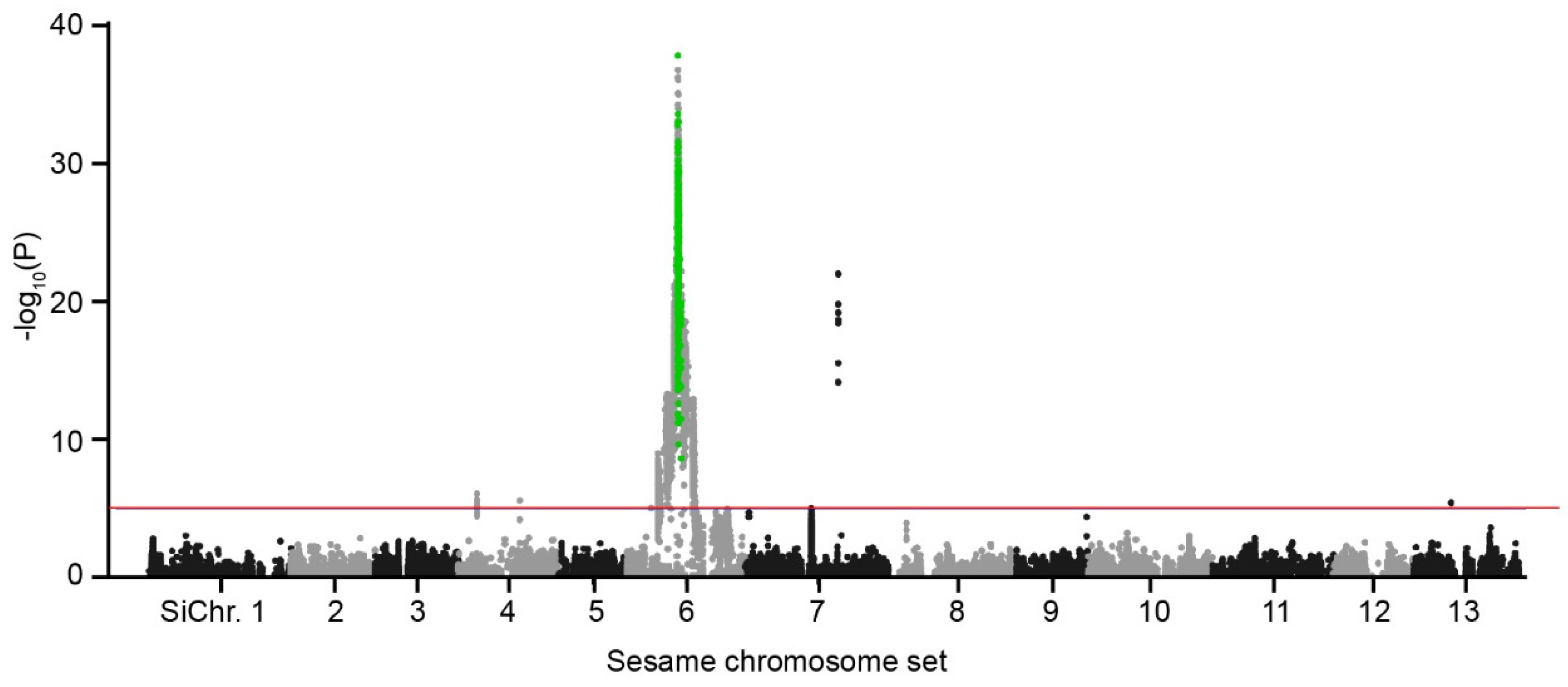
2.5. Sics1 Gene Identification
2.6. Structure Analysis of SiCS1 Gene and Homolog Comparison
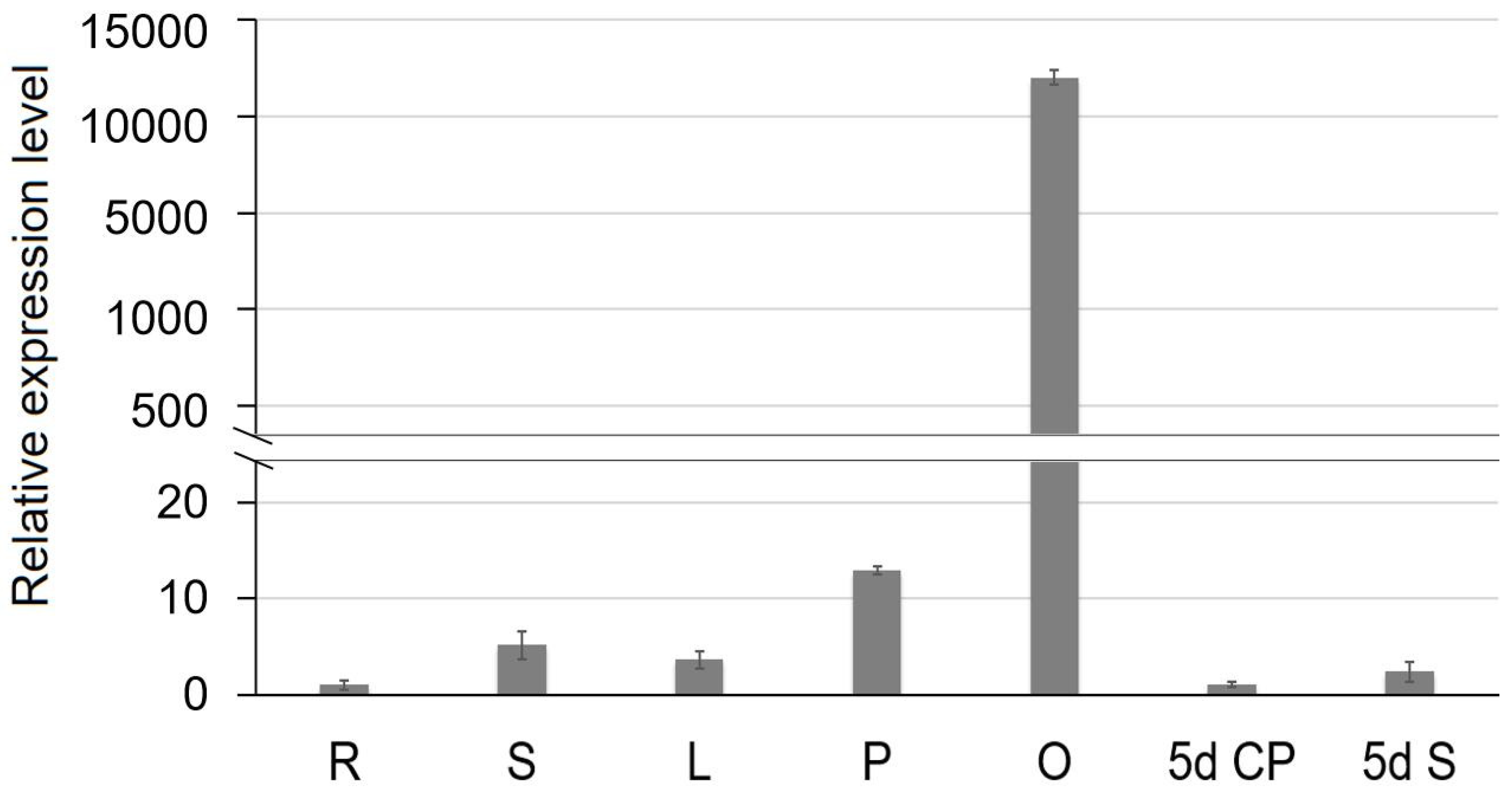
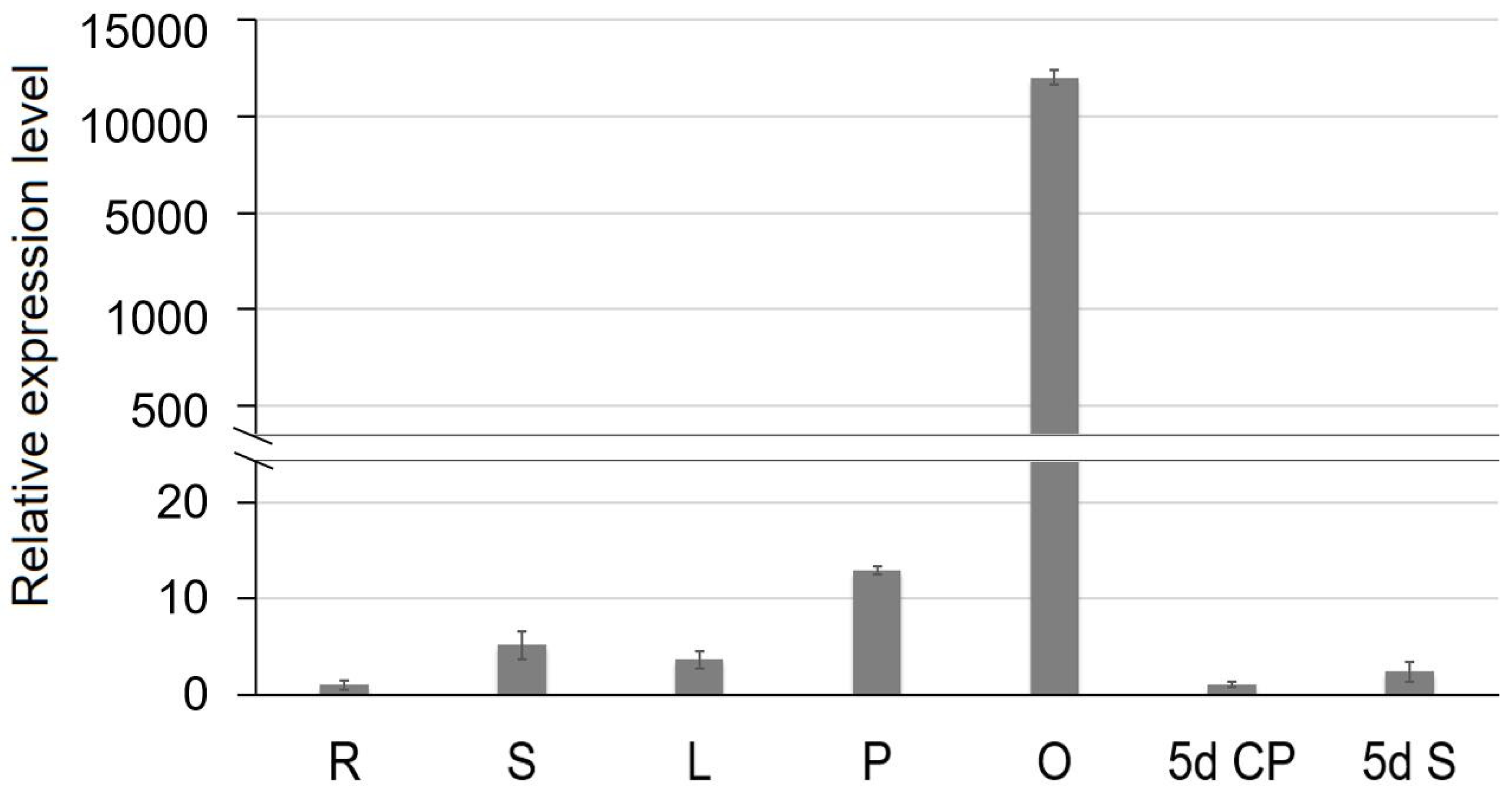
2.7. Expression Profiles of SiCS1 in Sesame
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Genomic DNA and RNA Extraction and Sequencing
4.3. Plant Hormone Determination
4.4. Sequencing Data Analysis
4.5. Statistical Significance and Candidate Gene Location
4.6. Cloning and Annotation of SiCS1 Gene
4.7. SiCS1 Homolog Detection and Phylogenetic Analysis
4.8. Expression Profile Assay of SiCS1 Gene
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AS-PCR | allele-specifific PCR |
| CKs | cytokinins |
| CL | capsule length |
| CRC | CRABS CLAW |
| cZ | cis-zeatin |
| czR | cis-zeatin riboside |
| DEGs | differentially expressed genes |
| DHZ | dihydrozeatin |
| EMS | Ehylmethane sulfonate |
| FC | fold change |
| FDR | false discovery rate |
| FKPM | the fragments per kilobase of transcript per million |
| GA | gibberellins |
| IAA | indole-3-acetic acid |
| InDel | Insertion-deletion |
| iP | isopenteny ladenine |
| iPR | isopenteny ladenosine |
| KEGG | Kyoto encyclopedia of genes and genomes |
| MT | mutant type |
| NGS | Next-generation sequencing technology |
| NR | Non- redundant |
| qRT-PCR | Quantitative real time PCR |
| QTL | Quantitative trait loci |
| RIL | Recombinant inbred line |
| SNC | Seed number per capsule |
| SNP | Single nucleotide polymorphism |
| tZ | trans-zeatin |
| tzR | trans-zeatin riboside |
| WT | wild type |
References
- Zhang, H.Y.; Miao, H.M.; Ju, M. Potential for adaption to climate change through genomic breeding in sesame. In Genomic Designing of Climate-Smart Oilseed Crops; Kole, C., Ed.; Springer Press: New York, NY, USA, 2019; pp. 371–440. [Google Scholar]
- Zhang, H.; Miao, H.; Wang, L.; Qu, L.; Liu, H.; Wang, Q.; Yue, M. Genome sequencing of the important oilseed crop Sesamum indicum L. Genome Boil. 2013, 14, 401. [Google Scholar]
- Sumathi, P.; Muralidharan, V. Inheritance of branching and important biometrical traits in sesame (Sesamum indicum L.). Indian J. Genet. 2009, 69, 1–5. [Google Scholar]
- Yol, E.; Uzun, B. Geographical Patterns of Sesame Accessions Grown Under Mediterranean Environmental Conditions, and Establishment of a Core Collection. Crop Sci. 2012, 52, 2206. [Google Scholar] [CrossRef]
- Diouf, M.; Boureima, S.; Diop, T. Gamma rays-induced mutant spectrum and frequency in sesame. Turk. J. Field Crops. 2010, 15, 99–105. [Google Scholar]
- Wu, K.; Liu, H.; Yang, M.; Tao, Y.; Ma, H.; Wu, W.; Zuo, Y.; Zhao, Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Boil. 2014, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Miao, H.; Li, C.; Wei, L.; Duan, Y.; Ma, Q.; Kong, J.; Xu, F.; Chang, S. Ultra-dense SNP genetic map construction and identification of SiDt gene controlling the determinate growth habit in Sesamum indicum L. Sci. Rep. 2016, 6, 31556. [Google Scholar] [CrossRef] [PubMed]
- Kamala, T. Gene action for seeds yield and yield components in sesame (Sesamum indicum L.). Indian J. Agric. Sci. 1999, 69, 773–774. [Google Scholar]
- Sumathi, P.; Muralidharan, V. Gene effects and inheritance of branching and other yield attributing characteristics in sesame (Sesamum indicum L.). Trop. Agric. Res. Ext. 2015, 16, 93. [Google Scholar] [CrossRef]
- Jatothu, J.L.; Dangi, K.S.; Kumar, S.S. Evaluation of sesame crosses for heterosis of yield and yield attributing traits. J. Trop Agr. 2013, 51, 84–91. [Google Scholar]
- Agrawal, M.M.; Singh, S.; Wawge, M.N.; Macwana, S.; Sasidharan, N. Correlation and path analysis for seed yield and yield attributing traits in Sesame germplasm (Sesamum indicum L.). Int. J. Chem. Stud. 2017, 5, 1099–1102. [Google Scholar]
- Krishna Devi, M.; Thirugnana, S.K.; Ganesan, J. Combining ability and heterosis for reproductive efficiency in sesame (Sesamum indicum L.). Sesame Safflower Newsl. 2003, 17, 5–9. [Google Scholar]
- Kumar, P.; Madhusudan, K.; Nadaf, H.L.; Patil, R.K.; Deshpande, S.K. Combining ability and gene action studies in inter-mutant hybrids of sesame (Sesamum indicum L.). Karnataka J. Agr. Sci. 2012, 25. [Google Scholar]
- Zhou, R.; Dossa, K.; Li, D.; Yu, J.; You, J.; Wei, X.; Zhang, X. Genome-Wide Association Studies of 39 Seed Yield-Related Traits in Sesame (Sesamum indicum L.). Int. J. Mol. Sci. 2018, 19, 2794. [Google Scholar] [CrossRef] [PubMed]
- Etherington, G.J.; Monaghan, J.; Zipfel, C.; MacLean, D. Mapping mutations in plant genomes with the user-friendly web application CandiSNP. Plant Methods 2014, 10, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Han, Y.; Li, Y.; Liu, D.; Sun, M.; Zhao, Y. Loci and candidate gene identification for resistance to Sclerotiniasclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J. 2015, 82, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhu, J.; Su, H.; Huang, M.; Wang, H.; Ding, S.; Zhang, B.; Luo, A.; Wei, S.; Tian, X.; et al. Bulked segregant RNA-seq reveals differential expression and SNPs of candidate genes associated with waterlogging tolerance in Maize. Front. Plant Sci. 2017, 8, 1022. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, Y.; Gao, J.; Ma, C.; Bi, Y. A comparative transcriptome analysis of a wild purple potato and its red mutant provides insight into the mechanism of anthocyanin transformation. PLoS ONE 2018, 13, e0191406. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhao, F.; Gao, S.; Wang, X.; Wei, A.; Chen, Z. Fine mapping of a male sterility gene ms-3 in a novel cucumber (cucumis sativus L.) mutant. Theor. Appl. Genet. 2018, 13, 1449. [Google Scholar] [CrossRef] [PubMed]
- Dilnur, T.; Peng, Z.; Pan, Z.; Palanga, K.K.; Jia, Y.; Gong, W.; Du, X. Association analysis of salt tolerance in Asiatic cotton (Gossypium arboretum) with SNP Markers. Int. J. Mol. Sci. 2019, 20, 2168. [Google Scholar] [CrossRef]
- Tan, C.; Liu, Z.; Huang, S.; Feng, H. Mapping of the male sterile mutant gene ftms in brassica rapa L. ssp. pekinensis via bsr-seq combined with whole-genome resequencing. Theor. Appl. Genet. 2019, 132, 355. [Google Scholar] [CrossRef]
- Sánchez-Martín, J.; Steuernagel, B.; Ghosh, S.; Herren, G.; Hurni, S.; Adamski, N. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 2016, 17, 221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Miao, H.M.; Wei, L.B.; Li, C.; Duan, Y.H.; Xu, F.F.; Qu, W.W.; Zhao, R.H.; Ju, M.; Chang, S.X. Identification of a SiCL1 gene controlling leaf curling and capsule indehiscence in sesame via cross-population association mapping and genomic variants screening. BMC Plant Biol. 2018, 18, 296. [Google Scholar] [CrossRef] [PubMed]
- Al Amin, G.M.; Kong, K.; Sharmin, R.A.; Kong, J.; Bhat, J.A.; Zhao, T. Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.). Int. J. Mol. Sci. 2019, 20, 2193. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shu, J.; Ali Mohamed, A.M.; Deng, X.; Zhi, X.; Bai, J.; Cui, Y.; Lu, X.; Du, Y.; Wang, X.; et al. Identification and Characterization of EI (Elongated Internode) Gene in Tomato (Solanum lycopersicum). Int. J. Mol. Sci. 2019, 20, 2204. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Miao, H.; Song, W.; Chen, C.; Zhang, H. Identification of sesame (Sesamum indicum L.) chromosomes using the BAC—FISH system. Plant Biol. 2018, 20, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Smyth, D.R. Crabs claw, a gene that regulates carpel and nectary development in arabidopsis, encodes a novel protein with zinc finger and helix-loop-helix domains. Development 1999, 126, 2387. [Google Scholar] [PubMed]
- Lee, J.Y.; Baum, S.F.; Alvarez, J.; Patel, A.; Chitwood, D.H.; Bowman, J.L. Activation of crabs claw in the nectaries and carpels of Arabidopsis. Plant Cell 2005, 17, 25–36. [Google Scholar] [CrossRef]
- Fourquin, C.; Vinaugerdouard, M.; Chambrier, P.; Bernededieu, A.; Scutt, C.P. Functional conservation between crabs claw orthologues from widely diverged angiosperms. Ann. Bot. 2007, 100, 651–657. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Huang, J.; Xu, Y.; Tanoi, K.; Ito, T. Fine-tuning of auxin homeostasis governs the transition from floral stem cell maintenance to gynoecium formation. Nat. Commun. 2017, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.; Smyth, D.R. Crabs claw and spatula genes regulate growth and pattern formation during gynoecium development in arabidopsis thaliana. Int. J. Plant Sci. 2002, 163, 17–41. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopaedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; TFennell, J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017. [Google Scholar] [CrossRef]
- Wei, L.B.; Miao, H.M.; Li, C.; Duan, Y.H.; Niu, J.J.; TZhang, D.; Zhao, Q.Y.; Zhang, H.Y. Development of SNP and InDel markers via de novo transcriptome assembly in Sesamum indicum L. Mol. Breed. 2014, 34, 2205–2217. [Google Scholar] [CrossRef]
- Liang, H.; Wang, C.; Li, Z.; Luo, X.Z.; Zou, G. Improvement of the silver-stained technique of polyacrylamide gel electrophoresis. Hereditas 2008, 30, 1379–1382. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Wei, L.B.; Miao, H.M.; Zhao, R.H.; Han, X.H.; Zhang, T.D.; Zhang, H.Y. Identification and testing of reference genes for Sesame gene expression analysis by quantitative real-time PCR. Planta 2013, 23, 7873–7889. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Type | CS Phenotype No. | cs Phenotype No. | Expected Ratio | χ2 Value |
|---|---|---|---|---|
| (sc1 × Yuzhi11) F1 | 42 | 0 | - | - |
| (Yuzhi11 × sc1) F1 | 46 | 0 | - | - |
| BC1 | 181 | 173 | 1:1 | 0.18 |
| F2 | 584 | 206 | 3:1 | 0.49 |
| Category | MT1 | MT2 | MT3 | WT1 | WT2 | WT3 |
|---|---|---|---|---|---|---|
| Obtained Clean Pair-end Reads (million) | 30.6 | 36.,3 | 31.7 | 32.5 | 35.3 | 43.9 |
| Obtained Clean Bases (Mbp) | 9159.9 | 10,873.4 | 9498.5 | 9749.9 | 10,588.4 | 13,147.7 |
| GC Content | 46.69% | 47.78% | 46.89% | 46.81% | 46.67% | 46.66% |
| Q30 % | 94.14% | 94.18% | 94.37% | 94.05% | 94.10% | 93.86% |
| Mapped Reads (million) (%) | 58.1 (94.81%) | 68.5 (94.29%) | 60.1 (94.72%) | 61.7 (94.78%) | 66.9 (94.71%) | 83.1 (94.70%) |
| Unique Mapped Reads (million) (%) | 56.9 (92.92%) | 66.1 (91.05%) | 58.9 (92.78%) | 60.5 (92.87%) | 65.6 (92.82%) | 81.3 (92.67%) |
| Multiple Map Reads (million) (%) | 1.2 (1.89%) | 2.4 (3.24%) | 1.2 (1.94%) | 1.2 (1.90%) | 1.3 (1.88%) | 1.8 (2.03%) |
| Sample Name | Raw Read Number (Million) | Coverage (×) a | Ratio of High-Quality Reads | Variant Loci Number | Unique LOCI c | |
|---|---|---|---|---|---|---|
| Q ≥ 20 | Q ≥ 30 | |||||
| Mutant cs1 (P1) | 54.2 | 22.97 | 96.12 | 91.65 | 284,753 | 124,578 |
| 15TP1783 (P2) | 57.6 | 24.39 | 95.90 | 91.35 | 360,602 | 200,427 |
| 131 F6 progeny b | 4999.3 | 2118.35 | 96.72 | 92.97 | / | / |
| Total | 5111.1 | 2165.72 | 96.71 | 92.95 | / | / |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; Li, C.; Duan, Y.; Qu, W.; Wang, H.; Miao, H.; Zhang, H. A SNP Mutation of SiCRC Regulates Seed Number Per Capsule and Capsule Length of cs1 Mutant in Sesame. Int. J. Mol. Sci. 2019, 20, 4056. https://doi.org/10.3390/ijms20164056
Wei L, Li C, Duan Y, Qu W, Wang H, Miao H, Zhang H. A SNP Mutation of SiCRC Regulates Seed Number Per Capsule and Capsule Length of cs1 Mutant in Sesame. International Journal of Molecular Sciences. 2019; 20(16):4056. https://doi.org/10.3390/ijms20164056
Chicago/Turabian StyleWei, Libin, Chun Li, Yinghui Duan, Wenwen Qu, Huili Wang, Hongmei Miao, and Haiyang Zhang. 2019. "A SNP Mutation of SiCRC Regulates Seed Number Per Capsule and Capsule Length of cs1 Mutant in Sesame" International Journal of Molecular Sciences 20, no. 16: 4056. https://doi.org/10.3390/ijms20164056
APA StyleWei, L., Li, C., Duan, Y., Qu, W., Wang, H., Miao, H., & Zhang, H. (2019). A SNP Mutation of SiCRC Regulates Seed Number Per Capsule and Capsule Length of cs1 Mutant in Sesame. International Journal of Molecular Sciences, 20(16), 4056. https://doi.org/10.3390/ijms20164056