1. Introduction
Tumors of the central nervous system (CNS) are a devastating disease. Of the 17,000 primary brain tumors diagnosed each year, approximately 60% are gliomas [
1], which arise in the brain from the supporting glial cells or their precursors [
2]. Gliomas in the brain typically are dangerous whether malignant or benign because of their location [
2]. Unfortunately, symptoms often do not present in patients until the tumor progresses, leading to late detection and poor prognosis [
3]. The World Health Organization (WHO) has graded gliomas from grades I-IV based on cell morphology, malignancy, and pathogenicity [
2]. Low grade gliomas are characterized by cellular morphology and cause local effects that do not spread in the brain. High grade gliomas are malignant and can spread throughout the brain tissue. Glioblastoma multiforme, or simply glioblastoma, (GBM; grade IV) account for 15% of all brain tumors and are classified as such due to their extensive differentiation, high invasiveness, and thus high malignancy of the tumor [
2,
3,
4]. The mean survival time of a patient from initial GBM diagnosis is approximately 15 months [
5] due to their highly invasive nature and late diagnosis after a patient experiences headaches, seizures, memory loss, vision changes, and personality changes. Current treatments for GBM include surgery, radiation, and chemotherapy [
3,
5] but these are practically ineffective.
One of the factors that increases glioma cell proliferation, motility, and invasiveness is autocrine/paracrine stimulation by cell adhesion molecule L1CAM (L1, CD171). L1 normally is expressed in developing neurons and is located on the axonal membrane, but it also is expressed in gliomas [
6,
7]. L1 has an extracellular ectodomain with five fibronectin domains and 6 immunoglobin-like domains, which is often excised and released in extracellular fluid. L1 interacts with various binding partners including itself, cell surface integrins, and other cellular surface components [
8]. L1 has a molecular weight of approximately 220 kDa and has an Arginine, Guanine and Aspartic acid (RGD) domain that binds to several integrin receptors [
9]. Two L1 binding partners that are particularly important for gliomas are integrins, which activate focal adhesion kinase (FAK), and fibroblast growth factor receptor (FGFR) [
10,
11]. Previously, our lab and others explored the interaction between L1 with these receptors along with the source of L1 expression [
11,
12,
13]. We have shown previously that L1-containing media and L1 ectodomain can increase migration of glioma cells [
12,
13,
14].
L1 also is present decorating the surface of minute exosomes, which are 40–100 nm membranous vesicles, released by glioma cells and formed by inward budding of the late endosomal multivesicular body membrane [
15]. Exosomes initially were isolated from blood samples and secreted by reticulocytes during differentiation [
16]. Tumor samples have been found to be rich in exosomes, where they induce tumor invasion and support tumor cell survival by promoting their invasive properties [
17,
18,
19,
20,
21]. We previously showed that glioma cells released exosomes decorated with L1 [
12], which raised the possibility that this, too, may be a source of L1 that could autocrine/paracrine stimulate glioma cell proliferation, motility, and invasion.
In this study, we aimed to determine if minute L1-decorated extracellular vesicles (exosomes) were capable of stimulating GBM cell motility, proliferation, and invasiveness. To do this, tumor cell exosomes, including those decorated with L1, were isolated from GBM cell culture media, and the effects of L1-decorated exosomes on glioma cell migration and proliferation were explored for their stimulatory activity. We evaluated the ability of L1-decorated exosomes to stimulate glioma cell motility and proliferation in vitro and invasion into embryonic chick brain tissue in vivo. We found that L1-decorated exosomes increased the migration and proliferation rates of glioma cells. We also used specific inhibitors against FAK and FGFR to implicate specific pathways leading to stimulation. Use of nanomolar concentrations of FAK and FGFR inhibitors resulted in a reduction in the L1-decorated exosomal stimulation of migration and proliferation rates back to below untreated control levels. Finally, we explored the ability of L1-decorated exosomes to promote invasiveness of glioma cells in vivo. There was an increased initial invasion of glioma cells when the L1-decorated exosomes were mixed with L1-attenuated glioma cells before injections, but this invasion did not continue far into brain tissue, indicating a sustained need for L1 stimulation of these cells for continuous invasion.
3. Discussion
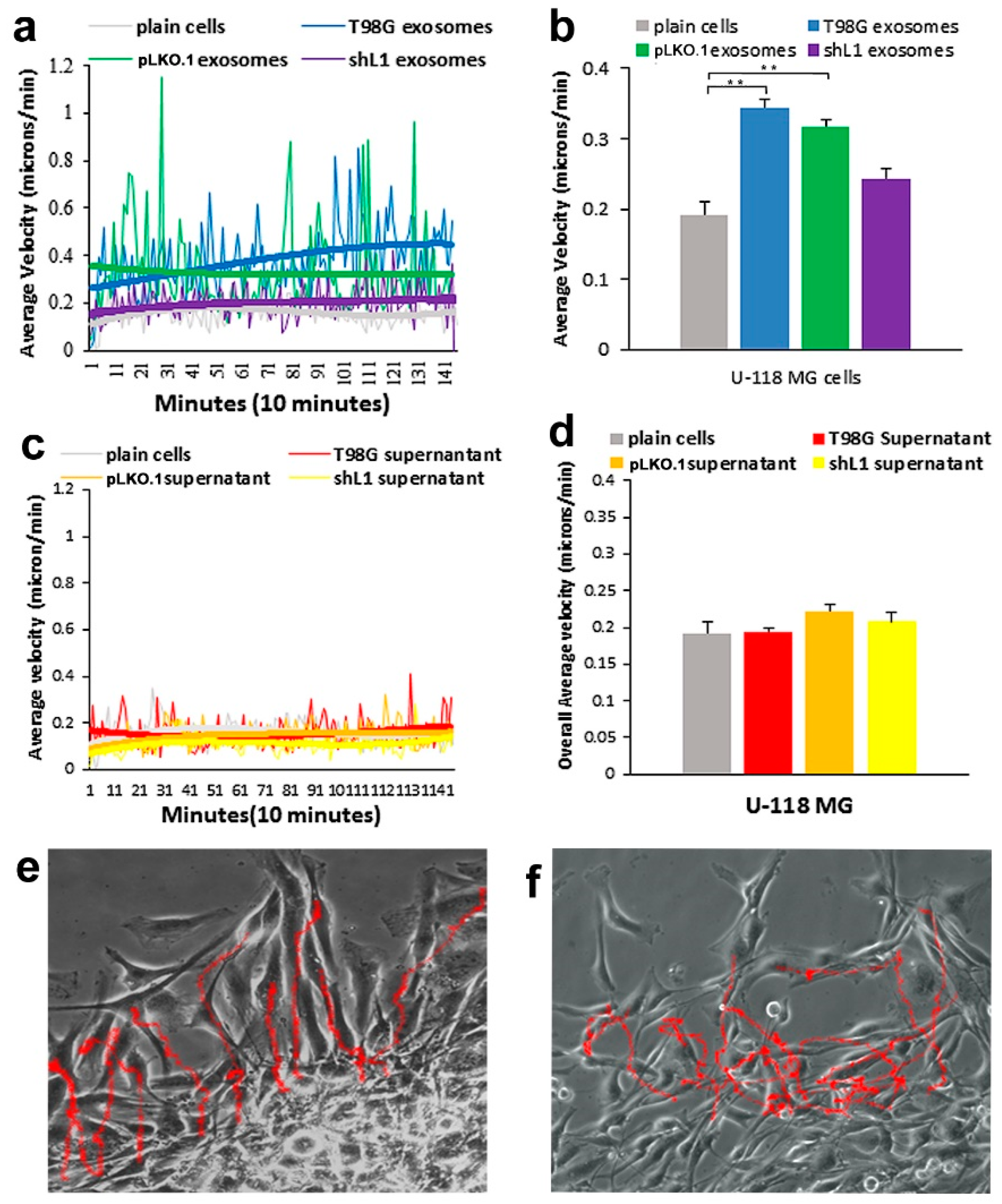
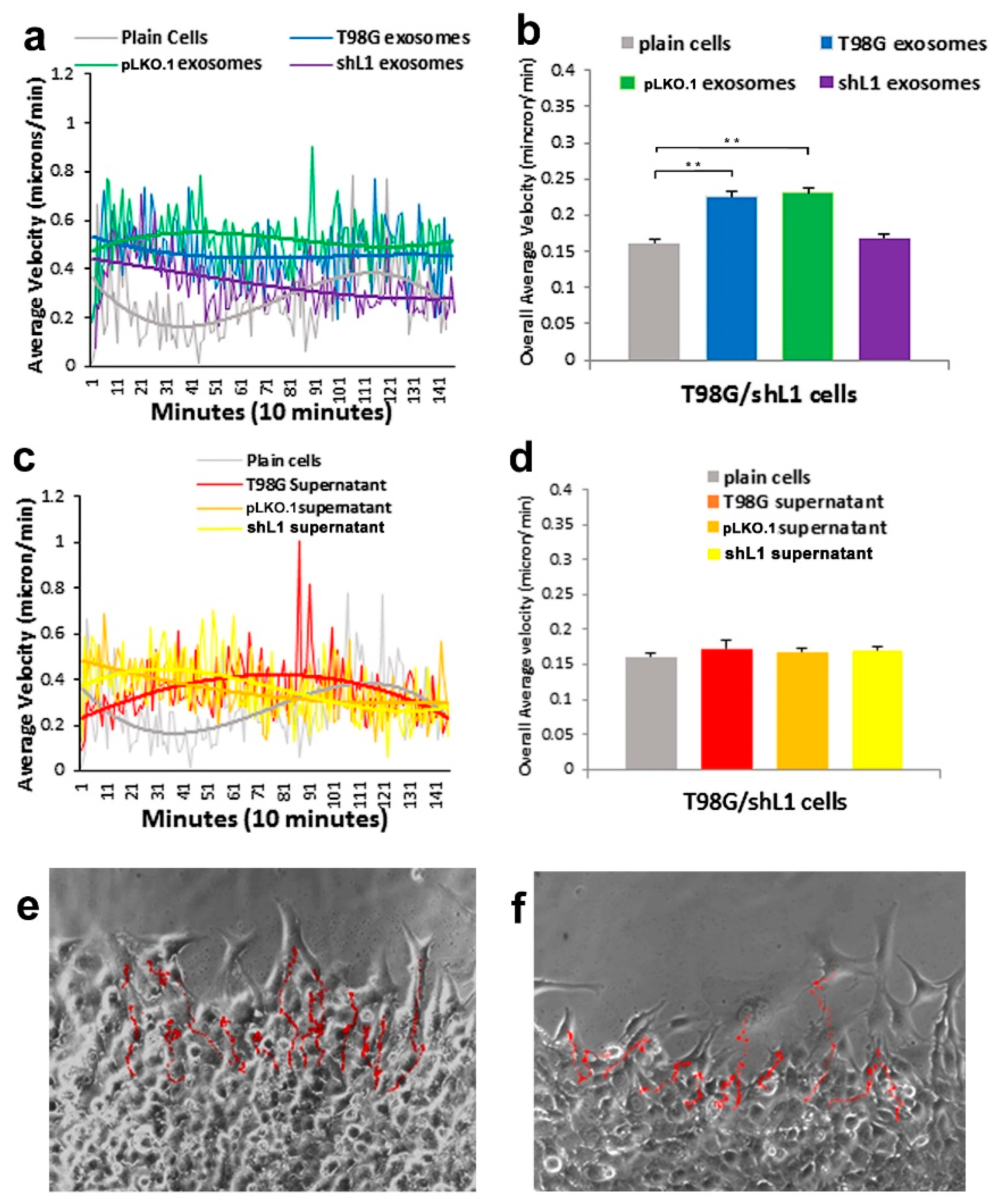
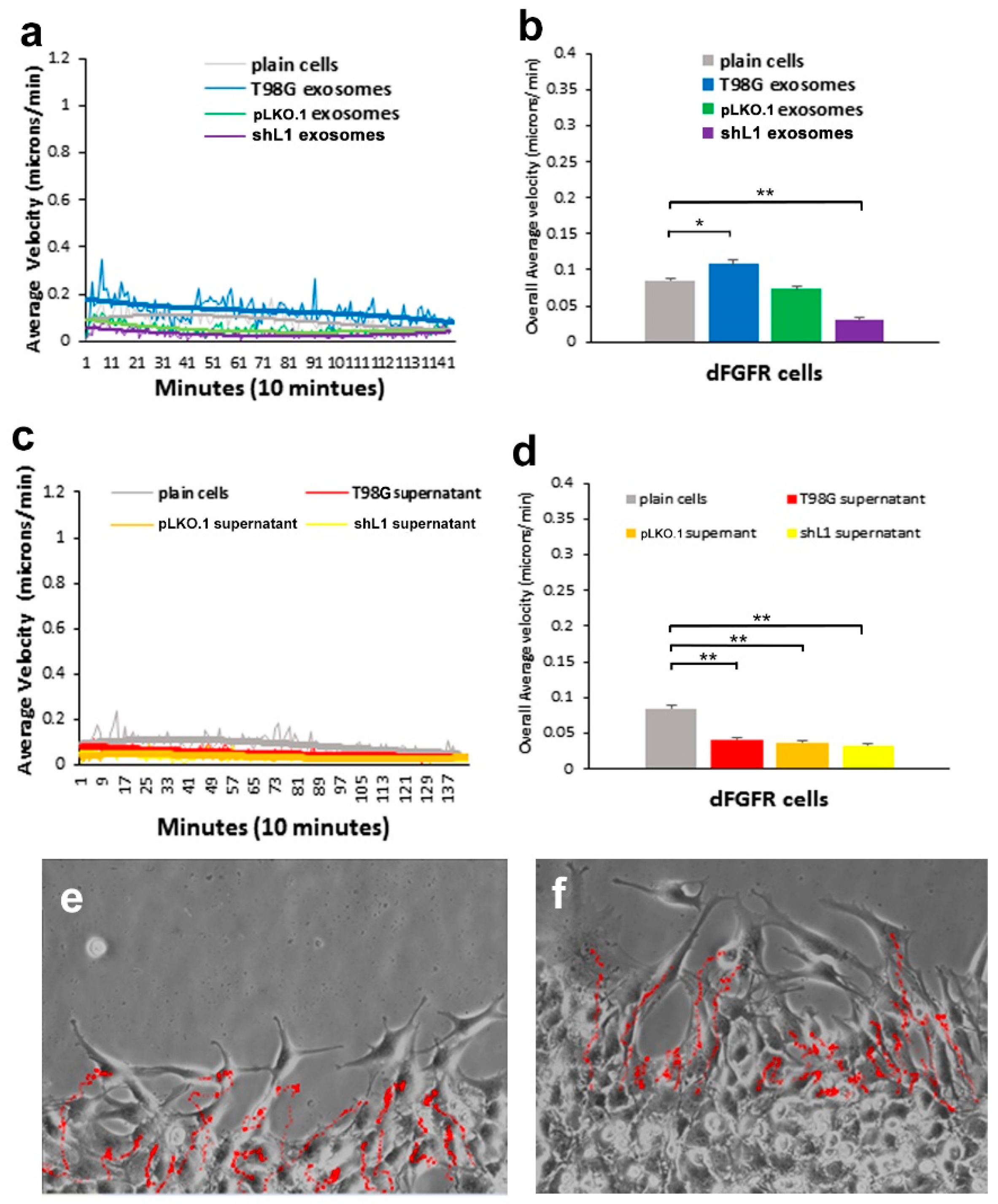
Glioblastoma (GBM) remains practically incurable despite multi-mode treatments that are given to patients (e.g., surgical resection, chemotherapy, radiation therapy) [
1,
2,
3,
4,
5]. This resistance to treatment is at least partly due to the high degree of motility and invasiveness of single cancer cells, which invade the brain tissue beyond the visible margins of the tumor. We previously identified the expression and release of L1 ectodomain as a potent autocrine/paracrine stimulator of motility, proliferation, and invasion for GBM cells [
12,
13,
14]. Here, that work was extended to show for the first time that L1-decorated exosomal vesicles released by GBM cells also are potent stimulators of motility and proliferation in vitro and invasion in vivo. When added to cultures, L1-decorated exosomes increased migration rates of U-118 MG, T98G, and T98G/dFGFR cells. In addition, L1-decorated exosomes also increased their proliferation, as indicated by both increased S phase and cell counts. To determine the extent to which exosomes initiated stimulatory signals through FGFRs and FAK, two potent small molecule inhibitors were used. The addition of FGFR and FAK inhibitors reduced the motility increase caused by exosomal addition back to basal levels. The chick brain invasion experiments showed that L1-decorated exosomes were capable of facilitating tissue invasion by GBM cells, thus demonstrating their importance in vivo. These results taken together demonstrate the importance of exosomal L1 for stimulation of GBM cells and the effectiveness of small molecule inhibitors in abrogating that stimulation.
Our previous results have shown that migration rates of GBM cells were increased by either when L1 ectodomain-containing media was added to L1 deficient cells [
12], or when L1 ectodomain was expressed by lentiviral transduction [
13]. The work presented here highlights the ability of L1-decorated exosomes from GBM cells to induce a significant increase in GBM cell migration and proliferation. Thus, these two different forms of L1 (soluble ectodomain and L1-decorated exosomes) are capable of modulating the behavior of GBM cell motility, proliferation, and invasiveness. Although we showed exosomal markers and TEM images, one limitation of our study is that we have not demonstrated exclusion of non-exosomal membranes from our preparations. However, our methods would have excluded membrane vesicles larger than 0.2 microns, such as microvesicles, and other large membrane debris.
There have been previous studies that have looked at the roles of GBM exosomes, but these did not report that a single membrane protein could significantly alter cell behavior. In one study, researchers looked at the effect on migration using exosomes from U-87 MG cells, whereby the exosomes increased the migration of GBM cells when added to trans-well cultures [
24]. This increased migration might have been due to L1 decorating the exosomes, since we previously showed that U-87 MG cells release L1-decorated exosomes [
12]. In another relevant study [
25], Skog et al. found that addition of primary GBM tumor exosomes nearly doubled the proliferation of U-87 glioma cells in vitro, but they did not ascribe this increase to any particular mechanism or protein. Those tumor exosomes were found to have migration-promoting mRNAs, as well as other factors that led to additional migration of the glioma cells [
25]. Those results are similar to our increased proliferation results, which largely were due to L1.
Our lab demonstrated that the FGFR is involved in increased proliferation of GBM cells with L1 ectodomain stimulation [
13,
14]. Other researchers have suggested that the epidermal growth factor receptor (EGFR) expressed on the surface of exosomes may also contribute to glioma cell proliferation. Several studies reported EM images of their exosomes and have found that these exosomes contain mRNAs, protein receptors, DNA, and microRNAs [
26]. There have been attempts to use exosomes as biomarkers to determine the presence of disease and other cellular dysfunction [
27]. The EM images provided here are similar to those in other published work. Electron micrographs, however, lack information beyond exosomal size and shape, so several techniques have been used to better determine their structure and interaction with GBM cells. One study used atomic force microscopy to visualize the surface of a cell and the attached exosomes while preserving the 3-D structure [
26].
In addition to visualization of exosomes, some studies have investigated their uptake. In this work, we showed that the uptake of fluorescent L1-decorated vs. L1-deficient exosomes differed over a 9-h time course. Binding and uptake of exosomes was clearly evident by the increasing fluorescence of glioma cells over time. These results align with other work that has shown in vitro exosome uptake [
24]. Previous work also has shown that exosomes express heparin sulfate proteoglycans (HSPGs) [
24], which facilitate binding to cells, and that when HSPGs were inhibited, there was a reduction in exosome binding and uptake. Our results are interesting in that L1 appeared to decrease exosome binding and uptake, or at least slow it down, although these results are not definitive and may be the result of adding different amounts of exosomes (see Results). Others also have shown that exosomes can alter the gene expression of cells that take them up. Thus, our results potentially contribute to the repertoire of molecules that modulate uptake of exosomes, which influences the behavior of GBM cells in multiple ways.
To further investigate exosomal cellular signaling, two known signaling pathways were explored. The FGFR is a known L1 receptor [
28] that has been a target of glioma treatments since it was found to induce GBM when it interacts with Transforming Acidic Coiled Coil genes (TACC) [
29]. Our work focused on the exosomal L1-FGFR signaling. Previously, we showed that L1 stimulates motility and proliferation of GBM cells in culture via FGFRs [
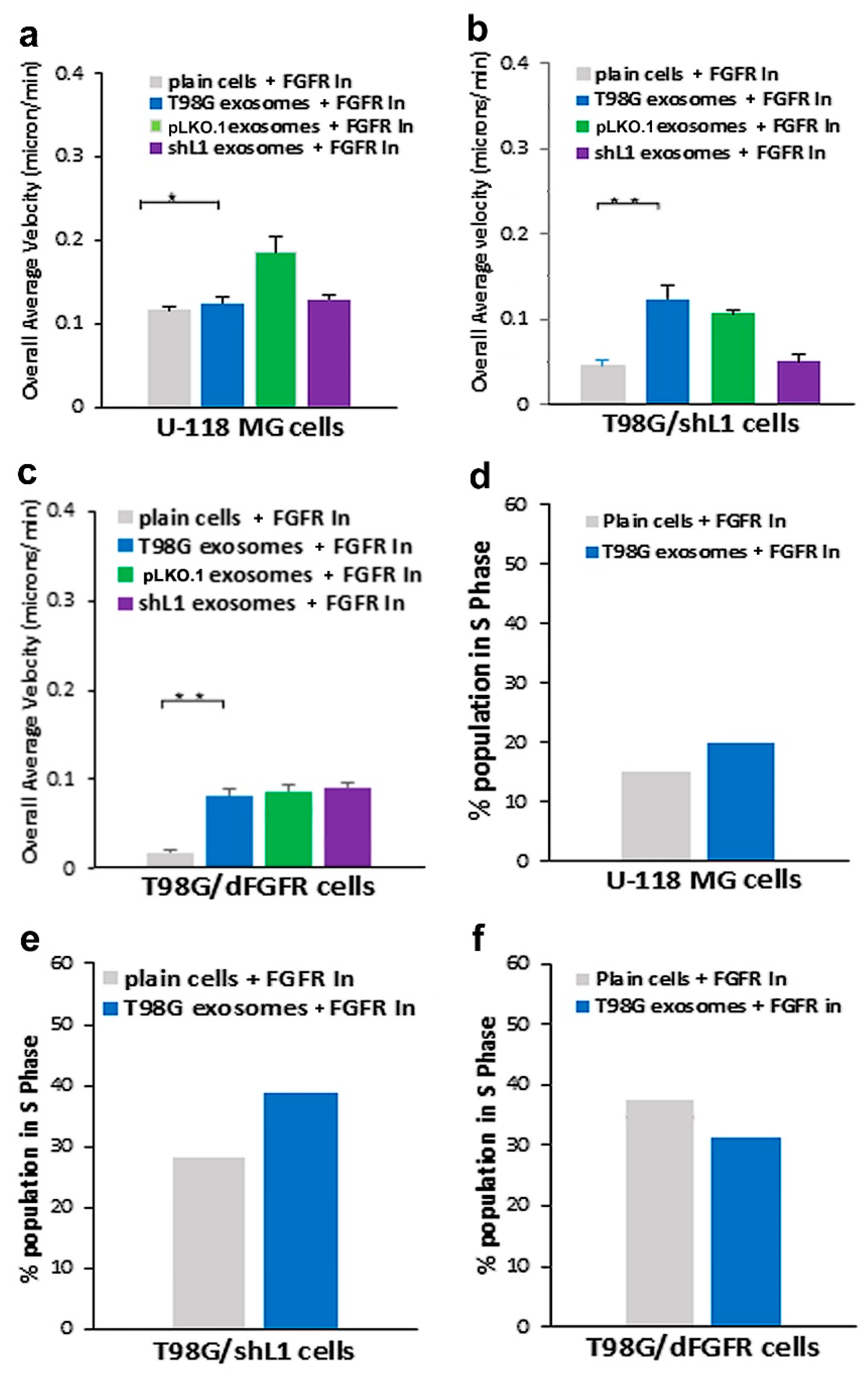
13]. The cell line with dominant-negative FGFR (T98G/dFGFR) showed decreased migration rates with soluble L1 ectodomain-conditioned media stimulation as compared to cells with normal FGFRs. This study extended that work to investigate the effect of L1-decorated exosomes on motility and proliferation via FGFRs. To reduce FGFR signaling, a potent inhibitor of FGFR (PD 173074) was used. When applied to cells in a
SuperScratch assay [
30], this FGFR inhibitor resulted in a marked reduction of the ability of exosomes to stimulate glioma motility. These motility rates went below those of plain T98G/shL1 cells, which indicates that other FGFR ligands (e.g., bFGF) may stimulate motility of T98G cells. Attenuation of FGFR signaling via the dominant-negative approach (T98G/dFGFR cells) resulted in the lowest motility rates seen in these studies. These findings support the idea that L1-decorated exosomes initiate sufficient signaling via the FGFR pathway to stimulate motility.
Another kinase explored in this work was FAK, which was presumed to become activated as a result of L1 stimulation of integrins. To explore the effect of L1-decorated exosomes on FAK signaling, a FAK inhibitor (PF 431396) was added to gliomas in a
SuperScratch assay. This FAK inhibitor significantly decreased the migration rate of glioma cell lines when added with exosomes. U-118 MG cell motility was reduced by about half compared to those stimulated with L1-decorated exosomes. The T98G/shL1 and T98G/dFGF cells showed significant reduction in velocity as well. L1-decorated exosomal stimulation of proliferation also was significantly reduced. This correlates with the published research findings that FAK inhibition reduces glioma progression and that FAK inhibition reduced tumor size of GBM, thus making this a viable clinical treatment for GBM [
31].
Although it was hypothesized by us that additional significant inhibition (e.g., additive effects) might be seen with the combination of both inhibitors, the results did not show much difference from either inhibitor alone. The addition of both inhibitors showed similar motility results to FGFR and FAK inhibitors individually for all GBM cell types used. This suggests that maximum motility inhibition had occurred because they share a common pathway, possibly FAK itself, as hypothesized by us previously for stimulation by L1 ectodomain [
14].
Our proliferation data showed that the addition of L1-decorated exosomes significantly increased cell cycle progression (the percentage of cells in S phase) of GBM cells. This is relevant to other studies that have investigated the effects of exosomes on proliferation. In one study, exosomes were isolated from gastric cancer cells and shown to promote proliferation through the PI3K/Akt and MAPK/ERK activation [
32]. In another study, exosomes from MDA-MB-231 cells were shown to promote proliferation of umbilical cord endothelial cells [
33]. In addition, exosomes from chronic myeloid leukemia and nasopharyngeal carcinoma tumors were used to promote proliferation of lymphocytes and other cells by increasing BCL-w, BCL-xl, and survivin, and a reduction of the pro-apoptotic molecules BAD, BAX and PUMA [
34]. Our results showing that L1-decorated exosomes promoted proliferation add another mechanism by which exosomes stimulate proliferation.
Based on the in vitro data that L1-decorated exosomes increased GBM cell motility, T98G/shL1 cells mixed with exosomes were injected into the optic tectum of developing chick embryos. Previously, it was shown that human and rat glioma cell lines could develop invasive tumors when injected into chick tectum [
35] and that L1 was necessary for invasion by T98G cells [
12]. Another in vivo study by others implicated L1 in GBM cell invasion by a correlative pattern of L1 immunostaining surrounding cells at the invasive front [
36]. In our work here, it is reported that L1-deficient T98G cells can form invasive tumors in chick brains only when mixed with L1-decorated exosomes, and not with L1-deficient exosomes. With L1-decorated exosomes, there was invasion into the optic tectum tissue beyond the generally smooth tumor margin, however, it was not as extensive as with normal T98G cells [
12]. We interpret this to reflect an initial stimulation by the exosomes, which was not sustained as the cells invaded into the tissue without further exogenous exosomal L1 stimulation. These data add to the idea that L1, as a component of exosomes, may be an important factor for GBM cell invasion in vivo. Therefore, L1 is a potential target for treatment, since patients that receive surgery have high recurrence of GBM. Since FAK and FGFR inhibitors used here were effective against L1-decorated exosomal stimulation, and they also were effective against L1 ectodomain stimulation [
14], they may be useful as treatments for GBM tumor cells that express, and are stimulated by, L1. However, our work here using two GBM cell lines (T98G and U-118 MG) shows that GBM cells that share the common characteristic of L1 stimulation may respond differently to such inhibitors.
4. Materials and Methods
4.1. Cell Lines and Cultures
Several cell lines and primary tissues were used for this work. For many experiments, two glioma cell lines, T98G and U-118 MG, were used. Both of these cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). T98G, a human WHO Grade IV glioma cell line [
37], expresses endogenous L1 [
12]. Another cell line was created (T98G/shL1) with a lentiviral vector insertion of a short hairpin RNA in T98G to reduce L1 expression (Open Biosystems TCTN0000063917; RHS3979-97052304) [
12]. The non-targeting control vector pLKO.1 with puromycin selection was used to create control T98G/pLKO.1 cell line. T98G/dFGFR cells [
13] are T98G cells that overexpress a dominant-negative FGFR1 construct that abrogates FGFR activity. U-118 MG, a human WHO Grade IV glioma line, does not express endogenous L1 protein [
12]. These two cell lines were used in comparison to the T98G lines to indicate endogenous vs exogenous L1 function. In addition, two surgical GBM samples of patient primary tumor cells from the Tissue Procurement Center of The Helen F. Graham Cancer Center and Research Institute of Christiana Hospital (Newark, DE) also were used in same experiments to compare cell lines to primary tumor sample to evaluate similarity to cultured lines. All tissues were cultured in Dulbecco’s Minimal Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 2 mM Penicillin/ Streptomycin (Pen/Strep) and 1 mM L-Glutamine (L-Glut). The tissues were incubated at 37° C with 5% CO
2.
4.2. Exosome Collection
Exosomes were isolated from T98G cell line culture media using ultracentrifugation. Cells were plated until 75% confluent in complete culture media (10% FBS). The complete media was removed from the plates and the cells were rinsed with Phosphate Buffer Saline (PBS). Next, DMEM containing 1% FBS, 2 mM Pen/Strep and L-Glut (low serum media), was added to cultured cells which were grown to 80–90% confluency for 24 h. This conditioned media was collected, centrifuged at low force (800 rpm) to remove any cells, and then supernatant was filtered using a 0.2 µm filters to remove any remaining cellular debris. This filtered media then was centrifuged at high speed for separation of exosomes from supernatant. The filtered media was centrifuged for 10 min at 1200× g and collecting supernatant, then 20 min at 10,000× g collecting supernatant, and finally 22 h at 100,000× g in an ultracentrifuge using a swinging bucket rotor. After this final centrifugation, the supernatant fraction (devoid of exosomes) and pellet fraction (containing exosomes) were used in experiments including motility and proliferation. These exosome pellets were gently rinsed with PBS, suspended in low serum (0.5–1% FBS) media, and added to cultured cells as needed. Typically, the exosomes isolated from a 10 cm culture were resuspended in 30–50 µL of low serum media. Some exosomes were used immediately, while others were stored for up to 30 days at 4 °C for later use.
4.3. SDS PAGE and Western Blotting
Cells on culture dishes were rinsed with phosphate buffered saline (PBS) and then solubilized in RIPA lysis buffer with Complete Mini Protease Inhibitor (Roche) for 3–5 min on ice. Cells were scraped, lysed by sonication and centrifuged to collect the cell lysates. Protein quantification was performed using the Bradford Assay (Sigma). Exosome pellets were dissolved in 100 μL RIPA lysis buffer with protease inhibitor for use in western blotting. 20 μg of protein from cell lysate and 50μL of exosome fraction were used to load lanes on the gel. Samples were prepared using NuPAGE LDS Sample Buffer (NP0007) and NuPAGE Sample Reducing Agent (NP0009) and were electrophoresed on Novex WedgeWell 4–20% Tris-Glycine Gel (XP04202BOX, Invitrogen) along with PageRuler Plus Prestained Protein Ladder (#26619, Thermo Scientific) in MOPS Running Buffer (BF-1131-1000, Baltimore Bioworks) at 120 VDC. The gel was transferred onto PVDF membrane at 4 °C at 30 VDC overnight. 5% nonfat dry milk dissolved in PBS with 0.01% Tween 20 used as the blocking reagent. Blots were incubated in 1:1000 NCAM-L1 antibody (UJ127.11, sc-53386, #B2117, Santa Cruz Biotechnology) or 1:1000 GAPDH antibody (0411, sc-47724, #D0419, Santa Cruz Biotechnology) with 1:1000 TSG 101 antibody (4A10, GTX70255, GeneTex) in 1% nonfat dry milk in PBS/Tween 20 for 1 h at room temperature. The blots were washed three times with PBS/Tween 20 for 10 min each and incubated with 1:50,000 HRP-conjugated secondary antibody (#115-035-062, Jackson ImmunoResearch) for 1 h. Finally, the blots were washed three times with PBS/Tween 20 and incubated with the chemiluminescent substrate (Lumigen ECL Ultra, TMA-6) for 2 min. The blots were imaged using BioRad ChemiDoc Imaging System.
4.4. Exosome Uptake
To determine exosome binding and uptake rates of cultured glioma cells, exosomes were labeled with fluorescent membrane dye DiO and were added to 50% confluent 10 cm plates of T98G/shL1 cells. Labeling of exosomes was done by adding 1 μL of Vybrant DiO (Invitrogen Molecular Probes cat# V22886) to 10 μL of exosomes for 30 min, followed by rinsing with PBS. T98G/shL1 cells in a 10 cm dish were incubated for 1–9 h with the labeled exosomes at 37 °C and then rinsed with PBS. Adherent cells were lightly trypsinized 0.05% trypsin/0.02% EDTA to suspend them, and cells were analyzed by flow cytometry to determine fluorescence of the cells using a Becton Dickinson FACSCalibur with 488 nm excitation and detection using the FL1 channel.
4.5. SuperScratch Assay
This method [
30] was used to evaluate the motility rates of cultured cells after a “wound” was made in confluent cell monolayers in 6-well plate wells (35 mm diameter). T98G/shL1 or U-118 cells were grown in complete media until about 75% confluency. A 1000 µL pipette tip was used to make a straight scratch through the cultured monolayer. Scratched monolayers were rinsed with PBS to remove debris and low serum media was added. Next, any additional treatments or exosomes were added to the plate. For
SuperScratch cell motility and cell cycle assays, 35 mm wells of cells were treated with either 3 mL of straight supernatant fraction or 5 μL of an exosome preparation diluted in 3 mL of fresh low serum media. Lids of 6-well plates were sealed with petroleum jelly on 3 sides to minimize evaporation and placed in the environmental chamber on our time-lapse microscope system built on a Nikon TE2000-E inverted microscope [
30]. MetaMorph software (Molecular Dynamics; ver.7.6.0.0) was used to control the microscope system and return to specific locations along the scratch edge to acquire images approximately every 10 min for a 24 h period using a Nikon 20× ELWD Plan Fluor phase contrast objective. After images were acquired, 15–20 cells per experimental condition were tracked from the collected images to determine the migration rate during 24 h cycle using the Track Points analysis function in MetaMorph. The average migration was calculated and used for migration rate as before [
12,
13,
14]. For example, when cells are tracked every 10 min over 20–24 h, this results in thousands of individual velocity values for each scratch edge (field of view), and each condition (e.g., U-118 MG cells + T98G exosomes) was analyzed in 2 or 3 fields of view in a single culture well. Then, data from the same conditions from several independent experiments were pooled to get the bar graph values of “overall average velocity,” and each bar may represent 6000–8000 or more averaged velocity values from several different fields of view and several different experiments.
4.6. Exosome Microscopy
To visualize exosome binding to cells, exosomes stained with lipophilic dyes FM 4–64 (Life technologies Molecular Probes cat# T-13320) or Vybrant DiO (Molecular Probes), or unstained exosomes, were added to T98G/shL1 cells in culture. To do this, each dye was added to the exosome fraction and incubated for 30 min at 37 °C. Excess dye was removed by washing exosomes with PBS twice and centrifugation to re-pellet exosomes. Live cells on coverslips were incubated with the stained exosomes for approximately 60 min at room temperature. Cells then were rinsed and fixed with 1% paraformaldehyde in PBS. Fixed cells incubated with attached exosomes were incubated with primary antibody against L1 (UJ127; cat# sc-53386, Santa Cruz Biotechnologies) for 1 h at room temperature and rinsed three times. Next, they were incubated with Alexa 488-conjugated secondary goat anti-mouse antibody (Jackson ImmunoResearch). Each preparation was stained briefly with 10 ug/mL bisbenzamide in PBS for nuclear staining. Coverslips were mounted on slides in a glycerol-based mounting medium, visualized, and photographed using a Nikon Plan Apo 60x oil immersion objective (N.A., 1.4) using the appropriate filter cube on a Nikon Microphot FX microscope and DXM1200 color CCD camera.
4.7. DNA Content/Cell Cycle and Cell Count Analyses
To evaluate the effects of the treatments on advancement through the cell cycle, each cell line was analyzed for DNA content using flow cytometry. Non-confluent (25%) cell cultures were trypsinized and transferred to a 15 mL tube in 2 mL of Soybean Trysin Inhibitor/DNase I. These tubes were centrifuged at 800 rpm and cell pellets were resuspended in a mixture of 500 µL PBS and 4.5 mL cold 70% ethanol and placed at −20 °C. After 4-7 days, the cells were pelleted, rinsed in PBS, and resuspended in 1 mL of DNA staining solution (200 µg/mL of DNase-free RNase A and 20 µg/mL propidium iodide in PBS). After the cells were incubated for 30 min at room temperature in the dark, they were analyzed for DNA fluorescence on a FACSCalibur flow cytometer using the FL2 channel. The percentage of cells in different cell cycle stages was determined from FL2-A data using ModFit LT software (Verity Software House; ver. 3.1). The results shown are from individual experiments. To supplement the FACS cell cycle analysis, cells were plated at 10% confluency and grown overnight. The next day, 10 spots were marked using an objective marker and counted for initial cell numbers within the circles. Next, the plates were washed twice with PBS. The appropriate treatment in low serum media was added to the plates and allowed to remain on the cells until the next day. 24 h later, the cells in the 10 marked circles were counted again. A cell count ratio was calculated by dividing the final counts by the initial counts. These values were graphed and used to determine any increases in cell division based upon treatment group. Cell cycle and cell count analyses were performed once.
4.8. FGFR and FAK Inhibition
To inhibit FGFR activity, cultures were treated with 50 nM PD173074 (Tocris), which is selective for FGFRs at this concentration (IC50 values are 5 and 21.5 nM for FGFR3 and FGFR1, respectively; Tocris website). To inhibit FAK activity, cultures were treated with 10 nM PF 431396 (Tocris), which also inhibits proline-rich tyrosine kinase 2 (PYK2) (IC50 values are 2 and 11 nM respectively; Tocris website).
4.9. TEM Imaging
To further characterize isolated exosomes, exosomes were resuspended in PBS and placed on 400 mesh formvar/carbon coated copper grids, washed with water and negatively stained with 2% uranyl acetate. Samples were imaged on a Zeiss Libra 120 transmission electron microscope operating at 120 kV, and images were acquired with a Gatan Ultrascan 1000 CCD camera.
4.10. In Vivo Invasion Analysis
The in vivo system used in this work was the White Leghorn chicken embryo. Fertilized eggs were obtained from the University of Delaware Department of Animal and Food Sciences. These embryos were incubated at 37.5 °C to embryonic day (E) 5. On E5, a window was cut into the egg and the air space membrane was removed. Approximately 10 µL of GBM cell suspension (~105 cells) was microinjected into the chick optic tectum using a PV830 pneumatic picopump (World Precision Instruments; Sarasota, FL). The GBM cell suspension contained cells, exosomes, low serum media, Fast Green FCF dye, and 30% Matrigel. For injections, 50 µL of T98G/shL1 or /pLKO.1 exosomes were added to the cell prep for injections. The total volume of injection sample was 430 µL (129 µl Matrigel + 50 µL shL1or pLKO.1 exosomes + 251 µL low serum DMEM). The total cell count was 4.3 × 106 cells in an injection sample (injection conc. is 107 cells/mL). After injection, 3 to 4 drops of sterile ampicillin solution (40 mg/mL) were added over the embryo. Transparent tape was placed over the window and the embryo was incubated until E9.
On E9, the embryos were sacrificed and dissected to obtain whole brains. These brains were fixed in 2% paraformaldehyde in PBS for 24–48 h. After fixation, the brains were embedded in embedding media composed of 3.5% agar and 8% sucrose in PBS. Next, the brains were sectioned at 300 microns using a Vibratome Series 1000 Sectioning System (Ted Pella, Redding, CA) for microscopic observation. These sections were mounted on slides in a buffered glycerol mounting media and sealed with nail polish. They were stored at in the dark before visualization using a Nikon Microphot FX epifluorescence microscope equipped with a Nikon DXM1200 color CCD camera. Tumors were scored as “attached” if there were continuous cells from the labeled tumor to the brain wall, which sometimes did not occur (i.e., some tumors grew in the ventricle unattached to the brain wall). Attached tumors were scored as “invasive” if fluorescent tumor cells visibly advanced beyond the generally smooth tumor margin of attachment into the surrounding chick brain tissue as single cells or groups of cells as before [
12]. Vibratome sections also were observed on a Nikon E800 epifluorescence microscope equipped with a Nikon C1 confocal scan head and 488 nm argon and 633 nm HeNe lasers, and confocal images were obtained using a 10× Plan Apo objective.
4.11. Statistical Analysis
To evaluate the data collected, all data were analyzed by Student’s T-test or ANOVA. After data was averaged for comparison of each treatment, these treatment groups averages were used to calculate standard deviation and standard error. These means were used in ANOVA to see if there was a difference in the treatment groups. The T-test was used to determine the significance of two samples that were compared when ANOVA found significance of treatment groups. All data analysis was performed in Microsoft Excel.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}