Sequence and Evolutionary Features for the Alternatively Spliced Exons of Eukaryotic Genes


Abstract
1. Introduction
2. Alternative Splicing and Exon Classification
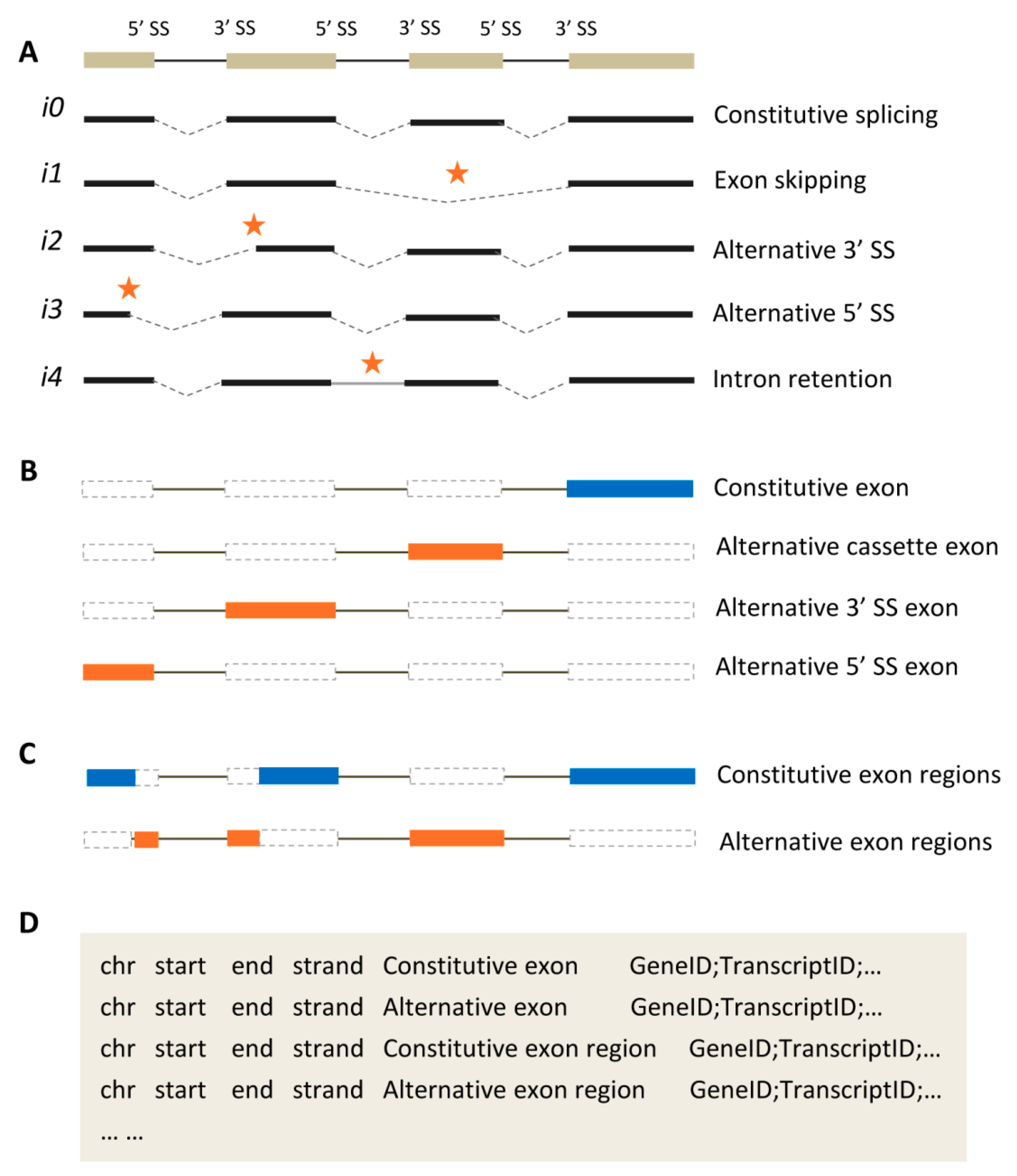
2.1. Representative Types of Alternative Splicing Events
2.2. Two Kinds of Exon Classification
2.3. Annotating Exons
3. Sequence Features of Alternative Exons
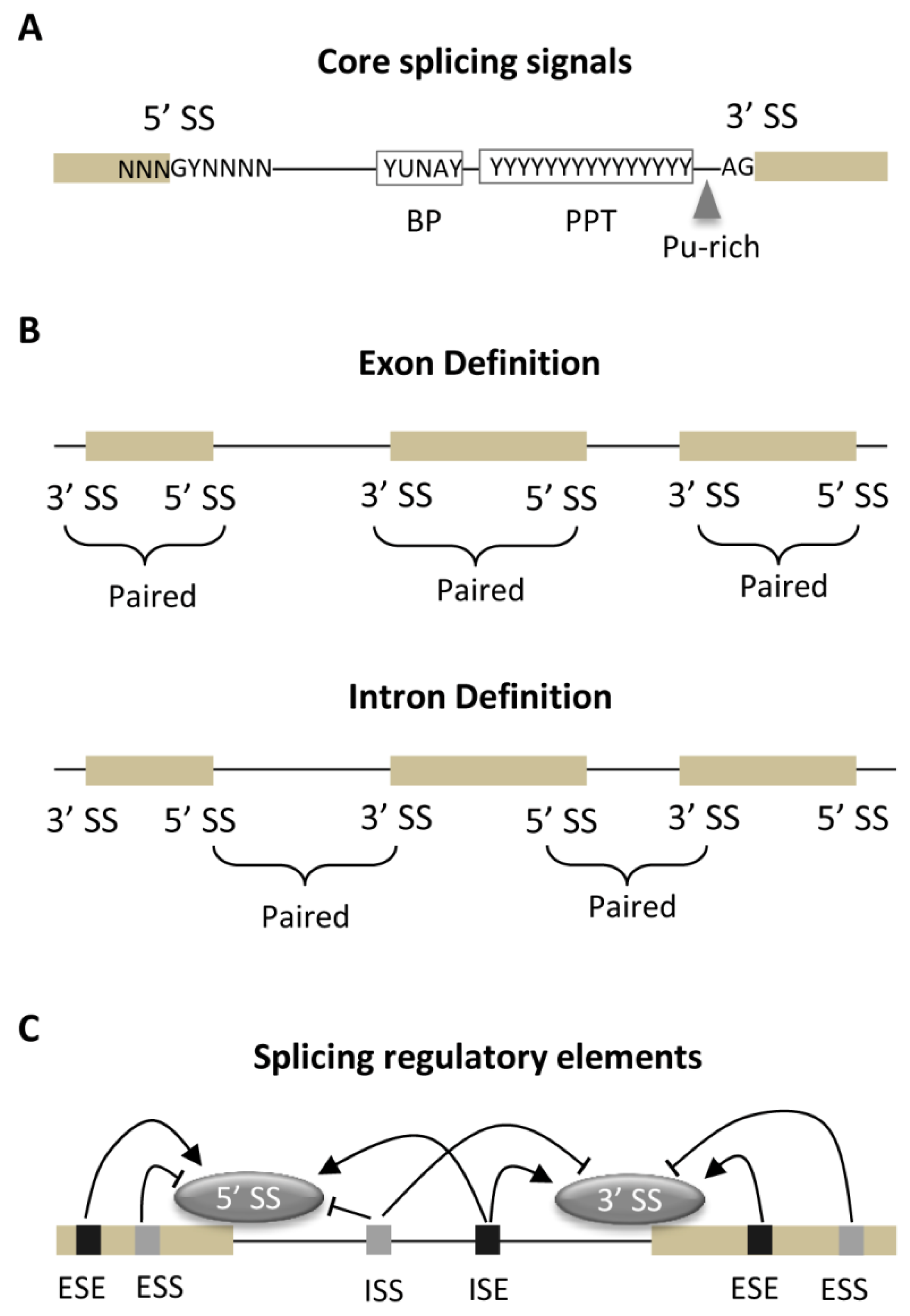
3.1. Core Splicing Signals and Regulatory Elements
3.2. Strength of Core Splicing Signals
3.3. Distribution of SREs
3.4. Exon-Intron Architecture
4. Evolutionary Features of Alternative Exons
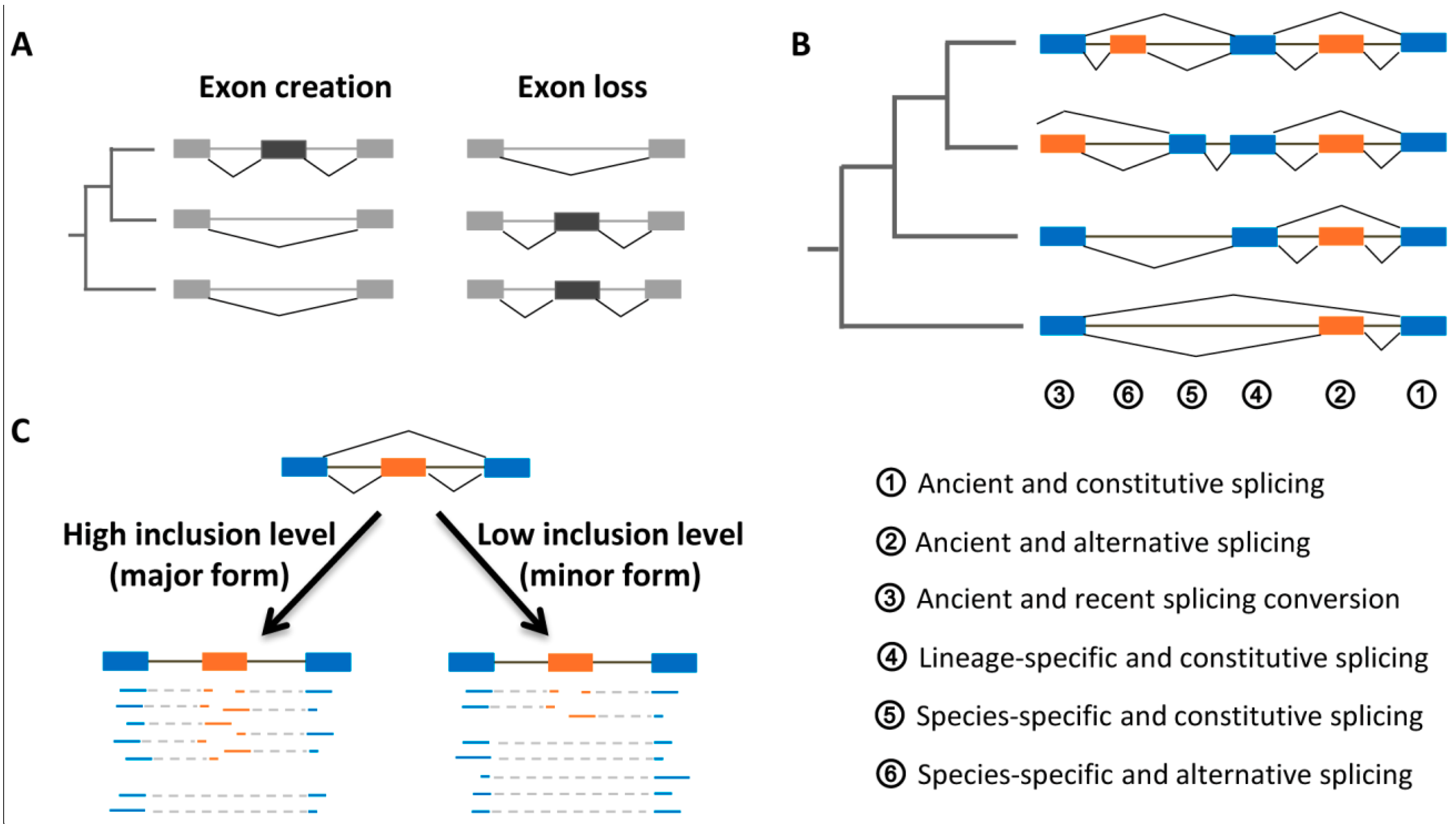
4.1. Evolutionary Ages and Inclusion Levels of Exons
4.2. Evolutionary Origins
4.3. Selective Constraints
4.4. Regulatory and Coding Roles
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kelly, D.E.; Hansen, M.E.B.; Tishkoff, S.A. Global variation in gene expression and the value of diverse sampling. Curr. Opin. Syst. Biol. 2017, 1, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Portin, P.; Wilkins, A. The evolving definition of the term “gene”. Genetics 2017, 205, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-M.; Xu, L.; Yau, M.Y.-C. Alternative mRNA splicing in the pathogenesis of obesity. Int. J. Mol. Sci. 2018, 19, E632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Jiang, Q.; Hao, H.; Ju, Z.; Yang, C.; Sun, Y.; Wang, C.; Zhong, J.; Huang, J.; et al. DNA methylation rather than single nucleotide polymorphisms regulates the production of an aberrant splice variant of IL6R in mastitic cows. Cell Stress Chaperones 2018, 23, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Cao, Y.; Ma, L. Alternative splicing in plant genes: A means of regulating the environmental fitness of plants. Int. J. Mol. Sci. 2017, 18, E432. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Deveson, I.W.; Brunck, M.E.; Blackburn, J.; Tseng, E.; Hon, T.; Clark, T.A.; Clark, M.B.; Crawford, J.; Dinger, M.E.; Nielsen, L.K.; et al. Universal alternative splicing of noncoding exons. Cell. Syst. 2018, 6, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The expanding landscape of alternative splicing variation in human populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef]
- Brett, D.; Pospisil, H.; Valcárcel, J.; Reich, J.; Bork, P. Alternative splicing and genome complexity. Nat. Genet. 2001, 30, 29–30. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 2017, 42, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Rätsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Ye, W.; Su, Y.; Chen, M.; Huang, G.; Wu, X. AStrap: Identification of alternative splicing from transcript sequences without a reference genome. Bioinformatics 2018, 35, 2654–2656. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.-J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res. 2017, 6, 100. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Deng, F.; Jia, X.; Li, C.; Lai, S.-J. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep. 2017, 7, 7648. [Google Scholar] [CrossRef] [PubMed]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef]
- Lee, Y.J.; Wang, Q.; Rio, D.C. Coordinate regulation of alternative pre-mRNA splicing events by the human RNA chaperone proteins hnRNPA1 and DDX5. Genes Dev. 2018, 32, 1060–1074. [Google Scholar] [CrossRef]
- Papasaikas, P.; Valcárcel, J. The spliceosome: The ultimate RNA chaperone and sculptor. Trends Biochem. Sci. 2016, 41, 33–45. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Hang, J.; Finci, L.I.; Lei, J.; Shi, Y. An atomic structure of the human spliceosome. Cell 2017, 169, 918–929. [Google Scholar] [CrossRef]
- Bai, R.; Wan, R.; Yan, C.; Lei, J.; Shi, Y. Structures of the fully assembled Saccharomyces cerevisiae spliceosome before activation. Science 2018, 360, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Marquez, Y.; Höpfler, M.; Ayatollahi, Z.; Barta, A.; Kalyna, M. Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res. 2015, 25, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Lev-Maor, G.; Ast, G. The emergence of alternative 3′ and 5′ splice site exons from constitutive exons. PLoS Comput. Biol. 2007, 3, e95. [Google Scholar] [CrossRef]
- Chen, F.-C. Are all of the human exons alternatively spliced? Brief. Bioinform. 2013, 15, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Dror, G.; Sorek, R.; Shamir, R. Accurate identification of alternatively spliced exons using support vector machine. Bioinformatics 2004, 21, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Huang, Q.; Guo, J.; Li-Ling, J.; Chen, X.; Ma, F. Comparative component analysis of exons with different splicing frequencies. PLoS ONE 2009, 4, e5387. [Google Scholar] [CrossRef]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced applications of RNA sequencing and challenges. Bioinform. Biol. Insights 2015, 9, 29–46. [Google Scholar] [CrossRef]
- Wong, M.S.; Kinney, J.B.; Krainer, A.R. Quantitative activity profile and context dependence of all human 5′ splice sites. Mol. Cell 2018, 71, 1012–1026. [Google Scholar] [CrossRef]
- Sheth, N.; Roca, X.; Hastings, M.L.; Roeder, T.; Krainer, A.R.; Sachidanandam, R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006, 34, 3955–3967. [Google Scholar] [CrossRef]
- Lin, J.-H.; Tang, X.-Y.; Boulling, A.; Zou, W.-B.; Masson, E.; Fichou, Y.; Raud, L.; Le Tertre, M.; Deng, S.-J.; Berlivet, I.; et al. First estimate of the scale of canonical 5′ splice site GT>GC variants capable of generating wild-type transcripts. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Das, U.; Wang, B.; Xie, J. The matrices and constraints of GT/AG splice sites of more than 1000 species/lineages. Gene 2018, 660, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Von Voithenberg, L.V.; Sánchez-Rico, C.; Kang, H.-S.; Madl, T.; Zanier, K.; Barth, A.; Warner, L.R.; Sattler, M.; Lamb, D.C. Recognition of the 3′ splice site RNA by the U2AF heterodimer involves a dynamic population shift. Proc. Natl. Acad. Sci. USA 2016, 113, E7169–E7175. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, J.; Zhang, Q.; Guo, D. A heuristic model for computational prediction of human branch point sequence. BMC Bioinform. 2017, 18, 459. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Xie, J. Widespread separation of the polypyrimidine tract from 3′ AG by G tracts in association with alternative exons in Metazoa and Plants. Front. Genet. 2019, 9, 741. [Google Scholar] [CrossRef]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, S.; Burstein, D.; Penn, O.; Savchenko, A.; Amit, M.; Schwartz, S.; Pupko, T.; Ast, G. Changes in exon-intron structure during vertebrate evolution affect the splicing pattern of exons. Genome Res. 2012, 22, 35–50. [Google Scholar] [CrossRef]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef]
- Grodecká, L.; Buratti, E.; Freiberger, T. Mutations of pre-mRNA splicing regulatory elements: Are predictions moving forward to clinical diagnostics? Int. J. Mol. Sci. 2017, 18, 1668. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z. Systematical identification of splicing regulatory cis-elements and cognate trans-factors. Methods 2014, 65, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xing, Y. CLIP-seq analysis of multi-mapped reads discovers novel functional RNA regulatory sites in the human transcriptome. Nucleic Acids Res. 2017, 45, 9260–9271. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Humenik, J.; Liebhaber, S.A. A cytosine-rich splice-regulatory determinant enforces functional processing of the human α-globin gene transcript. Blood 2019, 133, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Badr, E.; ElHefnawi, M.; Heath, L.S. Computational identification of tissue-specific splicing regulatory elements in human genes from RNA-Seq data. PLoS ONE 2016, 11, e0166978. [Google Scholar] [CrossRef] [PubMed]
- Goldammer, G.; Neumann, A.; Strauch, M.; Müller-McNicoll, M.; Heyd, F.; Preußner, M. Characterization of cis-acting elements that control oscillating alternative splicing. RNA Biol. 2018, 15, 1081–1092. [Google Scholar] [CrossRef]
- Tang, R.; Prosser, D.O.; Love, D.R. Evaluation of bioinformatic programmes for the analysis of variants within splice site consensus regions. Adv. Bioinform. 2016, 2016, 5614058. [Google Scholar] [CrossRef]
- Xu, Z.-C.; Wang, P.; Qiu, W.-R.; Xiao, X. iSS-PC: Identifying splicing sites via physical-chemical properties using deep sparse auto-encoder. Sci. Rep. 2017, 7, 8222. [Google Scholar] [CrossRef]
- Bretschneider, H.; Gandhi, S.; Deshwar, A.G.; Zuberi, K.; Frey, B.J. COSSMO: Predicting competitive alternative splice site selection using deep learning. Bioinformatics 2018, 34, i429–i437. [Google Scholar] [CrossRef]
- Busch, A.; Hertel, K.J. Splicing predictions reliably classify different types of alternative splicing. RNA 2015, 21, 813–823. [Google Scholar] [CrossRef]
- Cui, Y.; Cai, M.; Stanley, H.E. Comparative analysis and classification of cassette exons and constitutive exons. Biomed Res. Int. 2017, 2017, 7323508. [Google Scholar] [CrossRef] [PubMed]
- Grau-Bové, X.; Ruiz-Trillo, I.; Irimia, M. Origin of exon skipping-rich transcriptomes in animals driven by evolution of gene architecture. Genome Biol. 2018, 19, 135. [Google Scholar] [CrossRef] [PubMed]
- Soemedi, R.; Cygan, K.J.; Rhine, C.L.; Wang, J.; Bulacan, C.; Yang, J.; Bayrak-Toydemir, P.; McDonald, J.; Fairbrother, W.G. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 2017, 49, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.W.; Van Nostrand, E.L.; Liang, T.Y. Discovery and analysis of evolutionarily conserved intronic splicing regulatory elements. PLoS Genet. 2007, 3, e85. [Google Scholar]
- Rosenberg, A.B.; Patwardhan, R.P.; Shendure, J.; Seelig, G. Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 2015, 163, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef]
- De Boer, M.; van Leeuwen, K.; Geissler, J.; Belohradsky, B.H.; Kuijpers, T.W.; Roos, D. Mutation in an exonic splicing enhancer site causing chronic granulomatous disease. Blood Cells Mol. Dis. 2017, 66, 50–57. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Jin, Y.; Dong, H.; Shi, Y.; Bian, L. Mutually exclusive alternative splicing of pre-mRNAs. Wiley Interdiscip. Rev. RNA 2018, 9, e1468. [Google Scholar] [CrossRef]
- Anczuków, O.; Akerman, M.; Cléry, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-regulated alternative splicing in breast cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Magen, A.; Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2006, 35, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Lev-Maor, G.; Goren, A.; Sela, N.; Kim, E.; Keren, H.; Doron-Faigenboim, A.; Leibman-Barak, S.; Pupko, T.; Ast, G. The “alternative” choice of constitutive exons throughout evolution. PLoS Genet. 2007, 3, e203. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Amit, M.; Donyo, M.; Hollander, D.; Goren, A.; Kim, E.; Gelfman, S.; Lev-Maor, G.; Burstein, D.; Schwartz, S.; Postolsky, B.; et al. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012, 1, 543–556. [Google Scholar] [CrossRef]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.-X.; Tokheim, C.J.; Miller, S.E.; Xing, Y. Species-specific exon loss in human transcriptomes. Mol. Biol. Evol. 2014, 32, 481–494. [Google Scholar] [CrossRef][Green Version]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Modrek, B.; Lee, C.J. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat. Genet. 2003, 34, 177–180. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, H.; Yang, S.; Yu, H.; Li, J.; Jiang, H.; Su, J.; Yang, L.; Zhang, J.; McDermott, J.; et al. Origin and evolution of new exons in rodents. Genome Res. 2005, 15, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F.; Chasin, L.A. Comparison of multiple vertebrate genomes reveals the birth and evolution of human exons. Proc. Natl. Acad. Sci. USA 2006, 103, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.V.; Kim, N.; Lee, C.J. Global analysis of exon creation versus loss and the role of alternative splicing in 17 vertebrate genomes. RNA 2007, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Merkin, J.J.; Chen, P.; Alexis, M.S.; Hautaniemi, S.K.; Burge, C.B. Origins and impacts of new mammalian exons. Cell Rep. 2015, 10, 1992–2005. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Jiang, X.; Ditsiou, A.; Gao, Y.; Sun, J.; Lowenstein, E.D.; Huang, S.; Khaitovich, P. Predominant patterns of splicing evolution on human, chimpanzee and macaque evolutionary lineages. Hum. Mol. Genet. 2018, 27, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Rotival, M.; Quach, H.; Quintana-Murci, L. Defining the genetic and evolutionary architecture of alternative splicing in response to infection. Nat. Commun. 2019, 10, 1671. [Google Scholar] [CrossRef]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef]
- Hyung, D.; Kim, J.; Cho, S.Y.; Park, C. ASpedia: A comprehensive encyclopedia of human alternative splicing. Nucleic Acids Res. 2017, 46, D58–D63. [Google Scholar] [CrossRef]
- Chen, F.-C.; Wang, S.-S.; Chen, C.-J.; Li, W.-H.; Chuang, T.-J. Alternatively and constitutively spliced exons are subject to different evolutionary forces. Mol. Biol. Evol. 2006, 23, 675–682. [Google Scholar] [CrossRef]
- Chen, F.-C.; Liao, B.-Y.; Pan, C.-L.; Lin, H.-Y.; Chang, A.Y.-F. Assessing determinants of exonic evolutionary rates in mammals. Mol. Biol. Evol. 2012, 29, 3121–3129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Plass, M.; Eyras, E. Differentiated evolutionary rates in alternative exons and the implications for splicing regulation. BMC Evol. Biol. 2006, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Gelly, J.-C.; Lin, H.-Y.; de Brevern, A.G.; Chuang, T.-J.; Chen, F.-C. Selective constraint on human pre-mRNA splicing by protein structural properties. Genome Biol. Evol. 2012, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, K. The distribution pattern of genetic variation in the transcript isoforms of the alternatively spliced protein-coding genes in the human genome. Mol. Biosyst. 2015, 11, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Mockenhaupt, S.; Makeyev, E.V. Non-coding functions of alternative pre-mRNA splicing in development. Semin. Cell Dev. Biol. 2015, 47, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Blechingberg, J.; Poulsen, A.S.A.; Kjølby, M.; Monti, G.; Allen, M.; Ivarsen, A.K.; Lincoln, S.J.; Thotakura, G.; Vægter, C.B.; Ertekin-Taner, N.; et al. An alternative transcript of the Alzheimer’s disease risk gene SORL1 encodes a truncated receptor. Neurobiol. Aging 2018, 71, 266. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Items | Summary Description | |
|---|---|---|
| Sequence features | Core splicing signals |
|
| Splicing regulatory elements (SREs) |
| |
| Exon-intron architecture |
| |
| Evolutionary features | Origin |
|
| Selective constraint |
| |
| Regulatory and coding roles |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-Y.; Li, C.; Jia, X.; Lai, S.-J. Sequence and Evolutionary Features for the Alternatively Spliced Exons of Eukaryotic Genes. Int. J. Mol. Sci. 2019, 20, 3834. https://doi.org/10.3390/ijms20153834
Chen S-Y, Li C, Jia X, Lai S-J. Sequence and Evolutionary Features for the Alternatively Spliced Exons of Eukaryotic Genes. International Journal of Molecular Sciences. 2019; 20(15):3834. https://doi.org/10.3390/ijms20153834
Chicago/Turabian StyleChen, Shi-Yi, Cao Li, Xianbo Jia, and Song-Jia Lai. 2019. "Sequence and Evolutionary Features for the Alternatively Spliced Exons of Eukaryotic Genes" International Journal of Molecular Sciences 20, no. 15: 3834. https://doi.org/10.3390/ijms20153834
APA StyleChen, S.-Y., Li, C., Jia, X., & Lai, S.-J. (2019). Sequence and Evolutionary Features for the Alternatively Spliced Exons of Eukaryotic Genes. International Journal of Molecular Sciences, 20(15), 3834. https://doi.org/10.3390/ijms20153834