The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
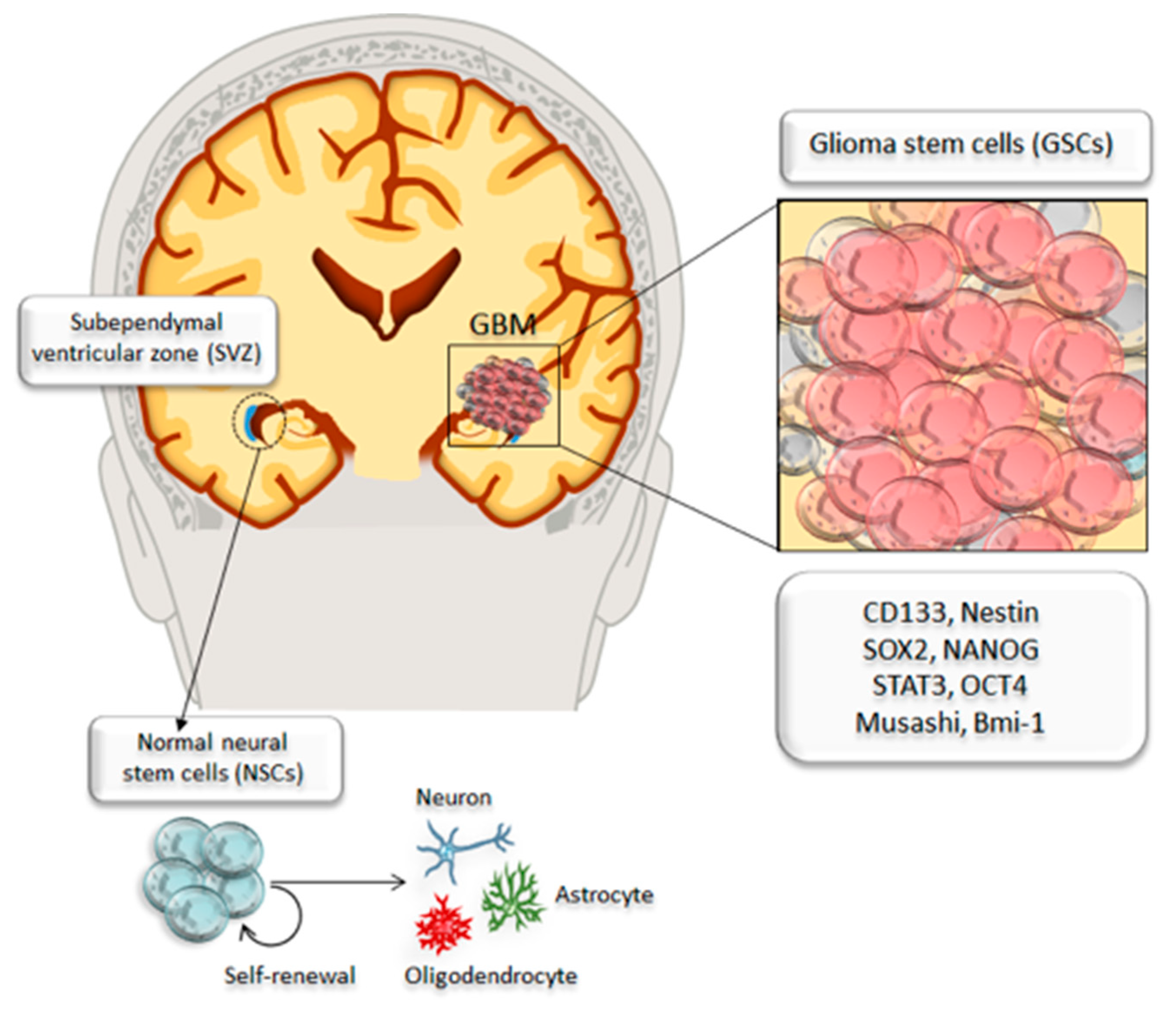
2. Identification and Targeting of Cancer Stem Cells in Glioblastoma
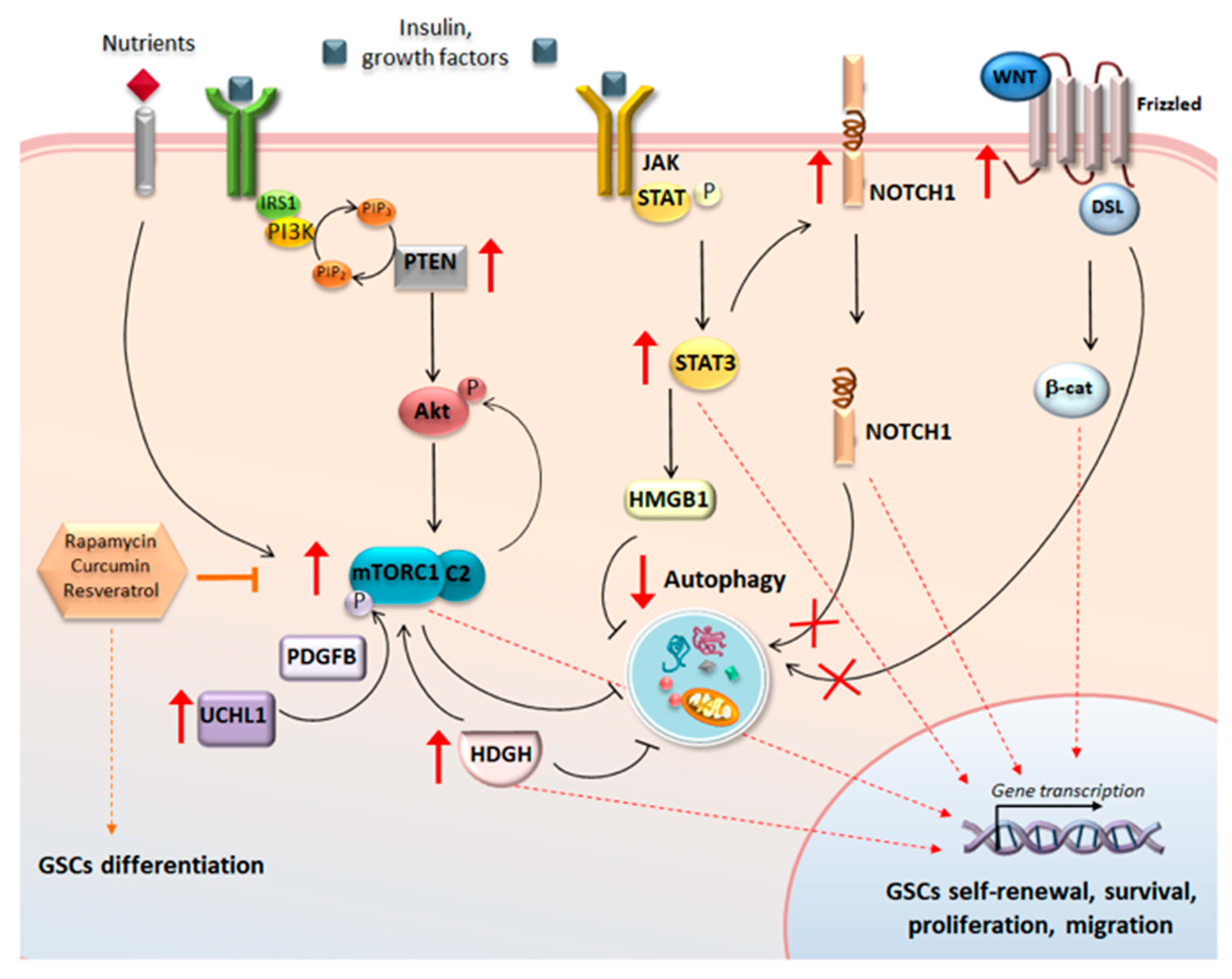
3. mTOR Function in Glioblastoma Cancer Stem Cells
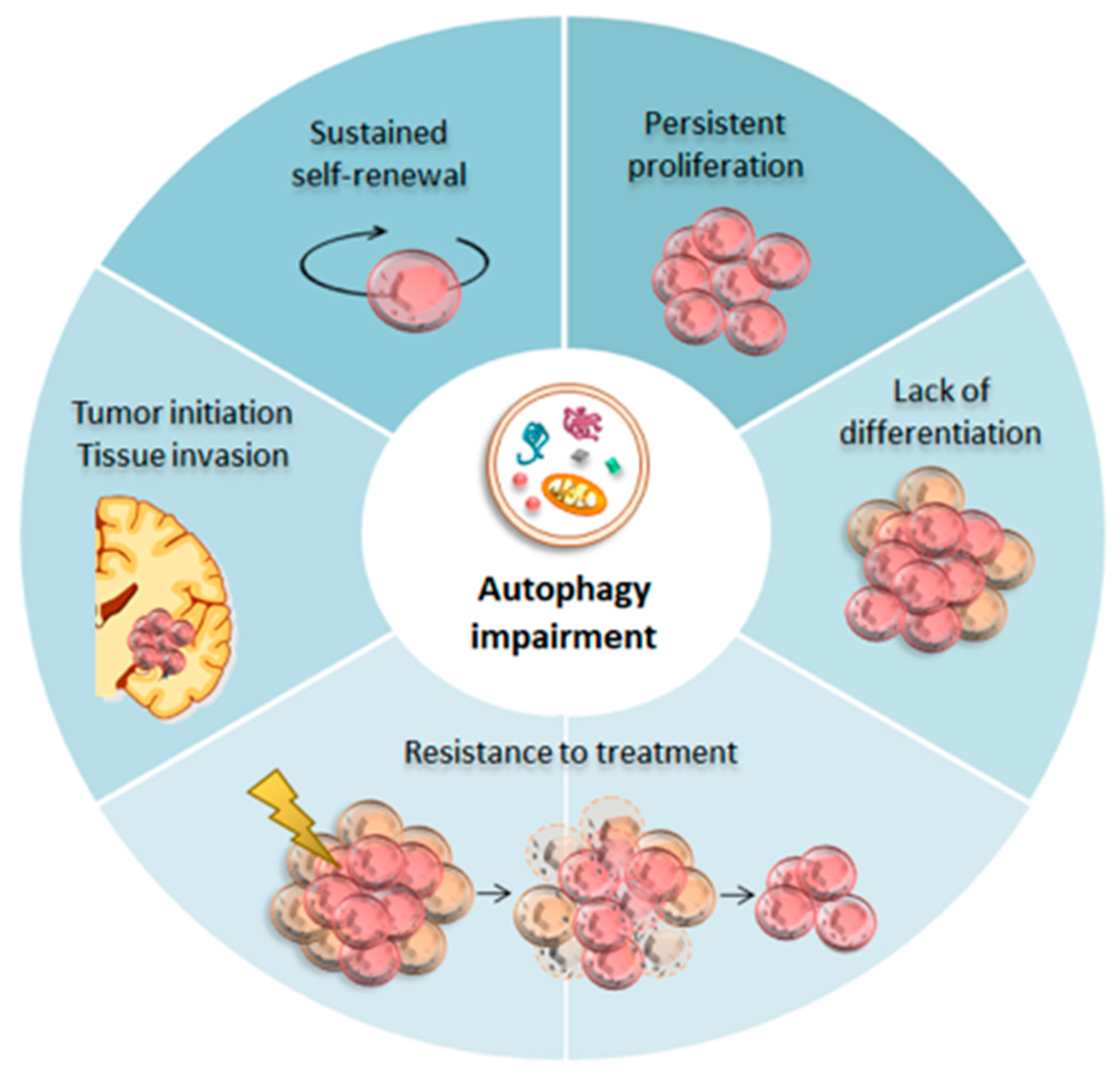
4. Autophagy in Glioblastoma Cancer Stem Cells
4.1. Autophagy Promoting GSCs
4.2. Autophagy Combating GSCs
5. The Cross-Talk between Autophagy and Glioblastoma Stem Cells-Controlling Pathways
5.1. Wnt/β-Catenin, Notch, and Autophagy in GSCs
5.2. UCHL1 and Autophagy in GSCs
5.3. SOX3, Hedgehog, and Autophagy in GSCs
5.4. STAT3 and Autophagy in GSCs
5.5. Epigenetic Enzymes and Autophagy in GSCs
6. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aml-1 | Acute myeloid leukemia 1 |
| BNIP3 | BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 |
| CNS | Central nervous system |
| CSCs | Cancer stem cells |
| EMT | Epithelial-mesenchymal transition |
| GSCs | Glioma stem-like cells |
| HDAC | Histone Deacetylases |
| mTOR | mammalian Target of Rapamycin |
| MDR | Multidrug resistance |
| MT1-MMP | Membrane Type-1 Matrix Metalloproteinase |
| NLGN3 | Neuroligin-3 |
| NSCs | Neural stem cells |
| PDGFB | Platelet-derived growth factor B |
| PI3K | Phosphoinositide 3-kinase |
| ROS | Reactive oxygen species (ROS) |
| SCP2 | Sterol carrier protein 2 |
| SGZ | Sugranular zone |
| SVZ | Subependymal ventricular zone |
| TFEB | Transcription factor EB |
| UCHL1 | ubiquitin carboxyl-terminal esterase L1 |
References
- Agnihotri, S.; Burrell, K.E.; Wolf, A.; Jalali, S.; Hawkins, C.; Rutka, J.T.; Zadeh, G. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Arch. Immunol. Ther. Exp. 2013, 61, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G.; ESMO Guidelines Working Group. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii93–iii101. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Choe, G.; Horvath, S.; Cloughesy, T.F.; Crosby, K.; Seligson, D.; Palotie, A.; Inge, L.; Smith, B.L.; Sawyers, C.L.; Mischel, P.S. Analysis of the phosphatidylinositol 3’-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003, 63, 2742–2746. [Google Scholar]
- Ermoian, R.P.; Kaprealian, T.; Lamborn, K.R.; Yang, X.; Jelluma, N.; Arvold, N.D.; Zeidman, R.; Berger, M.S.; Stokoe, D.; Haas-Kogan, D.A. Signal transduction molecules in gliomas of all grades. J. Neurooncol. 2009, 91, 19–26. [Google Scholar] [CrossRef]
- Yang, J.; Liao, D.; Wang, Z.; Liu, F.; Wu, G. Mammalian target of rapamycin signaling pathway contributes to glioma progression and patients’ prognosis. J. Surg. Res. 2011, 168, 97–102. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Levidou, G.; El-Habr, E.A.; Piperi, C.; Adamopoulos, C.; Samaras, V.; Boviatsis, E.; Thymara, I.; Trigka, E.A.; Sakellariou, S.; et al. Phosphorylated 4E-binding protein 1 (p-4E-BP1): A novel prognostic marker in human astrocytomas. Histopathology 2012, 61, 293–305. [Google Scholar] [CrossRef]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef]
- Hu, X.; Pandolfi, P.P.; Li, Y.; Koutcher, J.A.; Rosenblum, M.; Holland, E.C. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia 2005, 7, 356–368. [Google Scholar] [CrossRef][Green Version]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. NeuroOncology 2010, 12, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Weiss, W.A. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol. Biol. 2012, 821, 349–359. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhang, L.Q.; Zhang, X.G.; Li, X.; Ren, Y.B.; Ma, X.Y.; Li, X.G.; Wang, L.X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neurooncol. 2011, 103, 453–458. [Google Scholar] [CrossRef]
- Machado, L.E.; Alvarenga, A.W.; da Silva, F.F.; Roffé, M.; Begnami, M.D.; Torres, L.F.B.; da Cunha, I.W.; Martins, V.R.; Hajj, G.N.M. Overexpression of mTOR and p(240-244)S6 in IDH1 Wild-Type Human Glioblastomas Is Predictive of Low Survival. J. Histochem. Cytochem. 2018, 66, 403–414. [Google Scholar] [CrossRef]
- Neshat, M.S.; Mellinghoff, I.K.; Tran, C.; Stiles, B.; Thomas, G.; Petersen, R.; Frost, P.; Gibbons, J.J.; Wu, H.; Sawyers, C.L. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA 2001, 98, 10314–10319. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, E.; Ceruti, R.; Patton, V.; Russo, M.; Degrassi, A.; Croci, V.; Caprera, F.; Stortini, G.; Scanziani, E.; Pesenti, E.; et al. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histol. Histopathol. 2009, 24, 879–891. [Google Scholar]
- Iwamaru, A.; Kondo, Y.; Iwado, E.; Aoki, H.; Fujiwara, K.; Yokoyama, T.; Mills, G.B.; Kondo, S. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene 2007, 26, 1840–1851. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: Role of autophagic cell death. J. Natl. Cancer. Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef]
- Jiang, H.; White, E.J.; Conrad, C.; Gomez-Manzano, C.; Fueyo, J. Autophagy pathways in glioblastoma. Methods Enzymol. 2009, 453, 273–286. [Google Scholar]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. 2010, 17, 1515–1519. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Q.; Yang, J.; Lou, M.; Wang, A.; Dong, J.; Qin, Z.; Zhang, T. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res. 2010, 1313, 250–258. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z.Q. Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiating cells by inducing autophagy. Brain Res. 2011, 1371, 7–15. [Google Scholar] [CrossRef]
- Arcella, F.; Biagioni, M.; Oliva, A.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Liu, Q.; Nguyen, D.H.; Dong, Q.; Shitaku, P.; Chung, K.; Liu, O.Y.; Tso, J.L.; Liu, J.Y.; Konkankit, V.; Cloughesy, T.F.; et al. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J. Neurooncol. 2009, 94, 1–19. [Google Scholar] [CrossRef]
- Zhuang, W.; Qin, Z.; Liang, Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim. Biophys. Sin. 2009, 41, 341–351. [Google Scholar] [CrossRef]
- Toda, M. Glioma stem cells and immunotherapy for the treatment of malignant gliomas. ISRN Oncol. 2013, 2013, 673793. [Google Scholar] [CrossRef]
- Inda, M.M.; Bonavia, R.; Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef]
- Vessoni, A.T.; Muotri, A.R.; Okamoto, O.K. Autophagy in stem cell maintenance and differentiation. Stem Cells Dev. 2012, 21, 513–520. [Google Scholar] [CrossRef]
- Wang, C.; Liang, C.C.; Bian, Z.C.; Zhu, Y.; Guan, J.L. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 2013, 16, 532–542. [Google Scholar] [CrossRef]
- Chen, X.; He, Y.; Lu, F. Autophagy in Stem Cell Biology: A Perspective on Stem Cell Self-Renewal and Differentiation. Stem Cells Int. 2018, 2018, 9131397. [Google Scholar] [CrossRef]
- Tao, Z.; Li, T.; Ma, H.; Yang, Y.; Zhang, C.; Hai, L.; Liu, P.; Yuan, F.; Li, J.; Yi, L.; et al. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 2018, 9, 1063. [Google Scholar] [CrossRef]
- Lee, D.Y. Roles of mTOR Signaling in Brain Development. Exp. Neurobiol. 2015, 24, 177–185. [Google Scholar] [CrossRef]
- Casares-Crespo, L.; Calatayud-Baselga, I.; García-Corzo, L.; Mira, H. On the Role of Basal Autophagy in Adult Neural Stem Cells and Neurogenesis. Front. Cell. Neurosci. 2018, 12, 339. [Google Scholar] [CrossRef]
- LiCausi, F.; Hartman, N.W. Role of mTOR Complexes in Neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544. [Google Scholar] [CrossRef]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumor stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro Oncol. 2010, 12, 1205–1219. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Andreieva, S.V.; Korets, K.V.; Mykytenko, D.O.; Baklaushev, V.P.; Chekhonin, V.P.; Dmitrenko, V.V. mTOR inhibitor temsirolimus and MEK1/2 inhibitor U0126 promote chromosomal instability and cell type-dependent phenotype changes of glioblastoma cells. Gene 2016, 579, 58–68. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef]
- Galan-Moya, E.M.; LeGuelte, A.; Fernandes, E.L.; Thirant, C.; Dwyer, J.; Bidere, N.; Couraud, P.O.; Scott, M.G.; Junier, M.P.; Chneiweiss, H.; et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011, 12, 470–476. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Albert, L.; McKenna, E.; Karsy, M.; Rajdev, P.; Braun, A.; Murali, R. Deciphering the signaling pathways of cancer stem cells of glioblastoma multiforme: Role of Akt/mTOR and MAPK pathways. Adv. Enzyme Regul. 2011, 51, 164–170. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of glioblastoma multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210. [Google Scholar] [CrossRef]
- Fu, J.; Liu, Z.G.; Liu, X.M.; Chen, F.R.; Shi, H.L.; Pangjesse, C.S.; Ng, H.K.; Chen, Z.P. Glioblastoma stem cells resistant to temozolomide-induced autophagy. Chin. Med. J. (Engl). 2009, 122, 1255–1259. [Google Scholar]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of glioblastoma cells by mTOR inhibitors involves modulation of NFκB and PKC-α signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef]
- Gong, A.; Huang, S. FoxM1 and Wnt/β-catenin signaling in glioma stem cells. Cancer Res. 2012, 72, 5658–5662. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G., 2nd; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef]
- Sanai, N.; Tramontin, A.D.; Quinones-Hinojosa, A.; Barbaro, N.M.; Gupta, N.; Kunwar, S.; Lawton, M.T.; McDermott, M.W.; Parsa, A.T.; Manuel-García Verdugo, J.; et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 2004, 427, 740–744. [Google Scholar] [CrossRef]
- Paliouras, G.N.; Hamilton, L.K.; Aumont, A.; Joppé, S.E.; Barnabé-Heider, F.; Fernandes, K.J. Mammalian target of rapamycin signaling is a key regulator of the transit-amplifying progenitor pool in the adult and aging forebrain. J. Neurosci. 2012, 32, 15012–15026. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Basak, O.; Taylor, V. Stem cells of the mammalian brain. Cell. Mol. Life Sci. 2009, 66, 1057–1072. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring. Harb. Perspect Biol. 2015, 7, a020610. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.Ø.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–788. [Google Scholar] [CrossRef]
- Lendahl, U.; Zimmerman, L.B.; McKay, R.D. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60, 585–595. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008, 62, 505–514. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Ward, R.J.; Lee, L.; Graham, K.; Satkunendran, T.; Yoshikawa, K.; Ling, E.; Harper, L.; Austin, R.; Nieuwenhuis, E.; Clarke, I.D.; et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 2009, 69, 4682–4690. [Google Scholar] [CrossRef]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin α6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Baeza, N.; Colin, C.; Cayre, M.; Metellus, P.; Beclin, C.; Ouafik, L.; Figarella-Branger, D. A2B5 cells from human glioblastoma have cancer stem cell properties. Brain Pathol. 2010, 20, 211–221. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, Q.L.; Huang, Z.; Guryanova, O.A.; Huang, Q.A.; Shou, W.N.; Rich, J.N.; Bao, S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011, 30, 800–813. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef]
- Talsma, C.E.; Flack, C.G.; Zhu, T.; He, X.; Soules, M.; Heth, J.A.; Muraszko, K.; Fan, X. Oct4 regulates GBM neurosphere growth and its expression is associated with poor survival in GBM patients. Neuro Oncol. 2011, 13, 145–153. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Iacopino, F.; Angelucci, C.; Piacentini, R.; Biamonte, F.; Mangiola, A.; Maira, G.; Grassi, C.; Sica, G. Isolation of cancer stem cells from three human glioblastoma cell lines: Characterization of two selected clones. PLoS ONE 2014, 9, e105166. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Vega-Rubin-de-Celis, S.; Peña-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef]
- Pachow, D.; Wick, W.; Gutmann, D.H.; Mawrin, C. The mTOR signaling pathway as a treatment target for intracranial neoplasms. Neuro Oncol. 2015, 17, 189–199. [Google Scholar] [CrossRef]
- Tsang, C.K.; Qi, H.; Liu, L.F.; Zheng, X.F. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov. Today 2007, 12, 112–124. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. Mtor signaling in cancer and mtor inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Sato, A.; Sunayama, J.; Matsuda, K.; Tachibana, K.; Sakurada, K.; Tomiyama, A.; Kayama, T.; Kitanaka, C. Regulation of neural stem/progenitor cell maintenance by PI3K and mTOR. Neurosci. Lett. 2010, 470, 115–120. [Google Scholar] [CrossRef]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef]
- Ka, M.; Condorelli, G.; Woodgett, J.R.; Kim, W.Y. mTOR regulates brain morphogenesis by mediating GSK3 signaling. Development 2014, 141, 4076–4086. [Google Scholar] [CrossRef]
- Cloetta, D.; Thomanetz, V.; Baranek, C.; Lustenberger, R.M.; Lin, S.; Oliveri, F.; Atanasoski, S.; Ruegg, M.A. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci. 2013, 33, 7799–7810. [Google Scholar] [CrossRef]
- Lafourcade, C.A.; Lin, T.V.; Feliciano, D.M.; Zhang, L.; Hsieh, L.S.; Bordey, A. Rheb activation in subventricular zone progenitors leads to heterotopia, ectopic neuronal differentiation, and rapamycin-sensitive olfactory micronodules and dendrite hypertrophy of newborn neurons. J. Neurosci. 2013, 33, 2419–2431. [Google Scholar] [CrossRef]
- Kassai, H.; Sugaya, Y.; Noda, S.; Nakao, K.; Maeda, T.; Kano, M.; Aiba, A. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014, 7, 1626–1639. [Google Scholar] [CrossRef]
- Magini, A.; Polchi, A.; Di Meo, D.; Mariucci, G.; Sagini, K.; De Marco, F.; Cassano, T.; Giovagnoli, S.; Dolcetta, D.; Emiliani, C. TFEB activation restores migration ability to Tsc1-deficient adult neural stem/progenitor cells. Hum. Mol. Genet. 2017, 26, 3303–3312. [Google Scholar] [CrossRef]
- Magri, L.; Cambiaghi, M.; Cominelli, M.; Alfaro-Cervello, C.; Cursi, M.; Pala, M.; Bulfone, A.; García-Verdugo, J.M.; Leocani, L.; Minicucci, F.; et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 2011, 9, 447–462. [Google Scholar] [CrossRef]
- Nieto-González, J.L.; Gómez-Sánchez, L.; Mavillard, F.; Linares-Clemente, P.; Rivero, M.C.; Valenzuela-Villatoro, M.; Muñoz-Bravo, J.L.; Pardal, R.; Fernández-Chacón, R. Loss of postnatal quiescence of neural stem cells through mTOR activation upon genetic removal of cysteine string protein-α. Proc. Natl. Acad. Sci. USA 2019, 116, 8000–8009. [Google Scholar] [CrossRef]
- Bashir, T.; Cloninger, C.; Artinian, N.; Anderson, L.; Bernath, A.; Holmes, B.; Benavides-Serrato, A.; Sabha, N.; Nishimura, R.N.; Guha, A.; et al. Conditional astroglial Rictor overexpression induces malignant glioma in mice. PLoS ONE 2012, 7, e47741. [Google Scholar] [CrossRef]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef]
- Tee, A.R.; Sampson, J.R.; Pal, D.K.; Bateman, J.M. The role of mTOR signalling in neurogenesis, insights from tuberous sclerosis complex. Semin. Cell. Dev. Biol. 2016, 52, 12–20. [Google Scholar] [CrossRef]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef]
- Gangemi, R.M.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef]
- Garros-Regulez, L.; Garcia, I.; Carrasco-Garcia, E.; Lantero, A.; Aldaz, P.; Moreno-Cugnon, L.; Arrizabalaga, O.; Undabeitia, J.; Torres-Bayona, S.; Villanua, J.; et al. Targeting SOX2 as a Therapeutic Strategy in Glioblastoma. Front. Oncol. 2016, 6, 222. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Resveratrol promotes proteasome-dependent degradation of Nanog via p53 activation and induces differentiation of glioma stem cells. Stem Cell Res. 2013, 11, 601–610. [Google Scholar] [CrossRef]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Mammalian target of rapamycin inhibitors, temsirolimus and torin 1, attenuate stemness-associated properties and expression of mesenchymal markers promoted by phorbol-myristate-acetate and oncostatin-M in glioblastoma cells. Tumour Biol. 2017, 39, 1010428317695921. [Google Scholar] [CrossRef]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog and PI3K/Akt/mTOR pathways cooperate in suppressing survival, self-renewal and tumorigenic potential of glioblastoma-initiating cells. Mol. Cell. Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef]
- Mendiburu-Eliçabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Maciaczyk, D.; Dai, F.; Claus, R.; Firat, E.; Doostkam, S.; Bogiel, T.; Carro, M.S.; Döbrössy, M.; Herold-Mende, C.; et al. Resistance to hypoxia-induced, BNIP3-mediated cell death contributes to an increase in a CD133-positive cell population in human glioblastomas in vitro. J. Neuropathol. Exp. Neurol. 2012, 71, 1086–1099. [Google Scholar] [CrossRef]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘ménage-à-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef]
- Pratt, J.; Annabi, B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell Signal. 2014, 26, 917–924. [Google Scholar] [CrossRef]
- Natsumeda, M.; Maitani, K.; Liu, Y.; Miyahara, H.; Kaur, H.; Chu, Q.; Zhang, H.; Kahlert, U.D.; Eberhart, C.G. Targeting Notch Signaling and Autophagy Increases Cytotoxicity in Glioblastoma Neurospheres. Brain Pathol. 2016, 26, 713–723. [Google Scholar] [CrossRef]
- Bischof, J.; Westhoff, M.A.; Wagner, J.E.; Halatsch, M.E.; Trentmann, S.; Knippschild, U.; Wirtz, C.R.; Burster, T. Cancer stem cells: The potential role of autophagy, proteolysis, and cathepsins in glioblastoma stem cells. Tumour Biol. 2017, 39, 1010428317692227. [Google Scholar] [CrossRef]
- Xu, J.; Dang, Y.; Ren, Y.R.; Liu, J.O. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4764–4769. [Google Scholar] [CrossRef]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef]
- Friedman, M.D.; Jeevan, D.S.; Tobias, M.; Murali, R.; Jhanwar-Uniyal, M. Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid. Oncol. Rep. 2013, 30, 1645–1650. [Google Scholar] [CrossRef]
- Wang, W.J.; Long, L.M.; Yang, N.; Zhang, Q.Q.; Ji, W.J.; Zhao, J.H.; Qin, Z.H.; Wang, Z.; Chen, G.; Liang, Z.Q. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Li, J.J.; Zhou, Q.; Huang, A.; Liu, W.W.; Wang, K.; Gao, L.; Qi, S.T.; Lu, Y.T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11, 70. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtoli, L.; Miracco, C. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef]
- Coly, P.M.; Perzo, N.; Le Joncour, V.; Lecointre, C.; Schouft, M.T.; Desrues, L.; Tonon, M.C.; Wurtz, O.; Gandolfo, P.; Castel, H.; et al. Chemotactic G protein-coupled receptors control cell migration by repressing autophagosome biogenesis. Autophagy 2016, 12, 2344–2362. [Google Scholar] [CrossRef]
- Wang, C.C.; Hueng, D.Y.; Huang, A.F.; Chen, W.L.; Huang, S.M.; Yi-Hsin Chan, J. CD164 regulates proliferation, progression, and invasion of human glioblastoma cells. Oncotarget 2019, 10, 2041–2054. [Google Scholar] [CrossRef]
- Watt, S.M.; Chan, J.Y. CD164–a novel sialomucin on CD34+ cells. Leuk. Lymphoma 2000, 37, 1–25. [Google Scholar] [CrossRef]
- Tu, M.; Cai, L.; Zheng, W.; Su, Z.; Chen, Y.; Qi, S. CD164 regulates proliferation and apoptosis by targeting PTEN in human glioma. Mol. Med. Rep. 2017, 15, 1713–1721. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Hueng, D.Y.; Huang, H.Y.; Chen, J.Y.; Chen, Y. Magnolol and honokiol exert a synergistic anti-tumor effect through autophagy and apoptosis in human glioblastomas. Oncotarget 2016, 7, 29116–29130. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, H.; Liu, Y.; Guo, A.; Xu, X.; Qu, X.; Wang, S.; Zhao, J.; Li, Y.; Cao, Y. Resveratrol Inhibits the Invasion of Glioblastoma-Initiating Cells via Down-Regulation of the PI3K/Akt/NF-κB Signaling Pathway. Nutrients 2015, 7, 4383–4402. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, Y.; Sun, X.; He, X.; Wang, M.; Wang, Z.; Wang, Q.; Zhu, R.; Wang, S. Curcumin-Loaded Layered Double Hydroxide Nanoparticles-Induced Autophagy for Reducing Glioma Cell Migration and Invasion. J. Biomed Nanotechnol. 2016, 12, 2051–2062. [Google Scholar] [CrossRef]
- Filippi-Chiela, E.C.; Villodre, E.S.; Zamin, L.L.; Lenz, G. Autophagy interplay with apoptosis and cell cycle regulation in the growth inhibiting effect of resveratrol in glioma cells. PLoS ONE 2011, 6, e20849. [Google Scholar] [CrossRef]
- Xue, S.; Xiao-Hong, S.; Lin, S.; Jie, B.; Li-Li, W.; Jia-Yao, G.; Shun, S.; Pei-Nan, L.; Mo-Li, W.; Qian, W.; et al. Lumbar puncture-administered resveratrol inhibits STAT3 activation, enhancing autophagy and apoptosis in orthotopic rat glioblastomas. Oncotarget 2016, 7, 75790–75799. [Google Scholar] [CrossRef]
- Wang, J.; Qi, Q.; Feng, Z.; Zhang, X.; Huang, B.; Chen, A.; Prestegarden, L.; Li, X.; Wang, J. Berberine induces autophagy in glioblastoma by targeting the AMPK/mTOR/ULK1-pathway. Oncotarget 2016, 7, 66944–66958. [Google Scholar] [CrossRef]
- Hegazy, A.M.; Yamada, D.; Kobayashi, M.; Kohno, S.; Ueno, M.; Ali, M.A.; Ohta, K.; Tadokoro, Y.; Ino, Y.; Todo, T.; et al. Therapeutic strategy for targeting aggressive malignant gliomas by disrupting their energy balance. J. Biol. Chem. 2016, 291, 21496–21509. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef]
- Colella, B.; Faienza, F.; Carinci, M.; D’Alessandro, G.; Catalano, M.; Santoro, A.; Cecconi, F.; Limatola, C.; Di Bartolomeo, S. Autophagy induction impairs Wnt/β-catenin signalling through β-catenin relocalisation in glioblastoma cells. Cell Signal. 2019, 53, 357–364. [Google Scholar] [CrossRef]
- Sanchez-Diaz, P.C.; Chang, J.C.; Moses, E.S.; Dao, T.; Chen, Y.; Hung, J.Y. Ubiquitin carboxyl-terminal esterase L1 (UCHL1) is associated with stem-like cancer cell functions in pediatric high-grade glioma. PLoS ONE 2017, 12, e0176879. [Google Scholar] [CrossRef]
- Hussain, S.; Foreman, O.; Perkins, S.L.; Witzig, T.E.; Miles, R.R.; van Deursen, J.; Galardy, P.J. The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia 2010, 24, 1641–1655. [Google Scholar] [CrossRef]
- Hurst-Kennedy, J.; Chin, L.S.; Li, L. Ubiquitin C-terminal hydrolase l1 in tumorigenesis. Biochem. Res. Int. 2012, 2012, 123706. [Google Scholar] [CrossRef]
- Xiang, T.; Li, L.; Yin, X.; Yuan, C.; Tan, C.; Su, X.; Xiong, L.; Putti, T.C.; Oberst, M.; Kelly, K.; et al. The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PLoS ONE 2012, 7, e29783. [Google Scholar] [CrossRef]
- Hussain, S.; Feldman, A.L.; Das, C.; Ziesmer, S.C.; Ansell, S.M.; Galardy, P.J. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol. Cell Biol. 2013, 33, 1188–1197. [Google Scholar] [CrossRef]
- Yan, C.; Huo, H.; Yang, C.; Zhang, T.; Chu, Y.; Liu, Y. Ubiquitin C-Terminal Hydrolase L1 regulates autophagy by inhibiting autophagosome formation through its deubiquitinating enzyme activity. Biochem. Biophys. Res. Commun. 2018, 497, 726–733. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, L.; Niu, T.; Shao, L.; Li, H.; Wu, W.; Wang, W.; Lv, L.; Qin, Q.; Wang, F.; et al. Ubiquitin carboxyl terminal hydrolyase L1-suppressed autophagic degradation of p21WAF1/Cip1 as a novel feedback mechanism in the control of cardiac fibroblast proliferation. PLoS ONE 2014, 9, e94658. [Google Scholar] [CrossRef]
- Zhong, J.; Zhao, M.; Ma, Y.; Luo, Q.; Liu, J.; Wang, J.; Yuan, X.; Sang, J.; Huang, C. UCHL1 acts as a colorectal cancer oncogene via activation of the β-catenin/TCF pathway through its deubiquitinating activity. Int. J. Mol. Med. 2012, 30, 430–436. [Google Scholar] [CrossRef]
- Marjanovic Vicentic, J.; Drakulic, D.; Garcia, I.; Vukovic, V.; Aldaz, P.; Puskas, N.; Nikolic, I.; Tasic, G.; Raicevic, S.; Garros-Regulez, L.; et al. SOX3 can promote the malignant behavior of glioblastoma cells. Cell Oncol. (Dordr). 2019, 42, 41–54. [Google Scholar] [CrossRef]
- Zeng, X.; Ju, D. Hedgehog Signaling Pathway and Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 2279. [Google Scholar] [CrossRef]
- Maiti, S.; Mondal, S.; Satyavarapu, E.M.; Mandal, C. mTORC2 regulates hedgehog pathway activity by promoting stability to Gli2 protein and its nuclear translocation. Cell Death Dis. 2017, 8, e2926. [Google Scholar] [CrossRef]
- Cheng, W.; Zhang, C.; Ren, X.; Jiang, Y.; Han, S.; Liu, Y.; Cai, J.; Li, M.; Wang, K.; Liu, Y.; et al. Bioinformatic analyses reveal a distinct Notch activation induced by STAT3 phosphorylation in the mesenchymal subtype of glioblastoma. J. Neurosurg. 2017, 126, 249–259. [Google Scholar] [CrossRef]
- Liu, T.; Li, A.; Xu, Y.; Xin, Y. Momelotinib sensitizes glioblastoma cells to temozolomide by enhancement of autophagy via JAK2/STAT3 inhibition. Oncol. Rep. 2019, 41, 1883–1892. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, J.; Dui, D.; Ren, H.; Liu, J. High mobility group box 1 induces the activation of the Janus kinase 2 and signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway in pancreatic acinar cells in rats, while AG490 and rapamycin inhibit their activation. Bosn. J. Basic Med. Sci. 2016, 16, 307–312. [Google Scholar] [CrossRef]
- Marampon, F.; Leoni, F.; Mancini, A.; Pietrantoni, I.; Codenotti, S.; Letizia, F.; Megiorni, F.; Porro, G.; Galbiati, E.; Pozzi, P.; et al. Histone deacetylase inhibitor ITF2357 (givinostat) reverts transformed phenotype and counteracts stemness in in vitro and in vivo models of human glioblastoma. J. Cancer Res. Clin. Oncol. 2019, 145, 393–409. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an inhibitor of histone methyltransferase, induces autophagy-dependent differentiation of glioma stem-like cells. Sci. Rep. 2016, 6, 38723. [Google Scholar] [CrossRef]
- Ferrucci, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Gambardella, S.; Frati, A.; Fornai, F. Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia. Int. J. Mol. Sci. 2018, 19, 2756. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. https://doi.org/10.3390/ijms20153824
Ryskalin L, Gaglione A, Limanaqi F, Biagioni F, Familiari P, Frati A, Esposito V, Fornai F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. International Journal of Molecular Sciences. 2019; 20(15):3824. https://doi.org/10.3390/ijms20153824
Chicago/Turabian StyleRyskalin, Larisa, Anderson Gaglione, Fiona Limanaqi, Francesca Biagioni, Pietro Familiari, Alessandro Frati, Vincenzo Esposito, and Francesco Fornai. 2019. "The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies" International Journal of Molecular Sciences 20, no. 15: 3824. https://doi.org/10.3390/ijms20153824
APA StyleRyskalin, L., Gaglione, A., Limanaqi, F., Biagioni, F., Familiari, P., Frati, A., Esposito, V., & Fornai, F. (2019). The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. International Journal of Molecular Sciences, 20(15), 3824. https://doi.org/10.3390/ijms20153824