Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS?



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
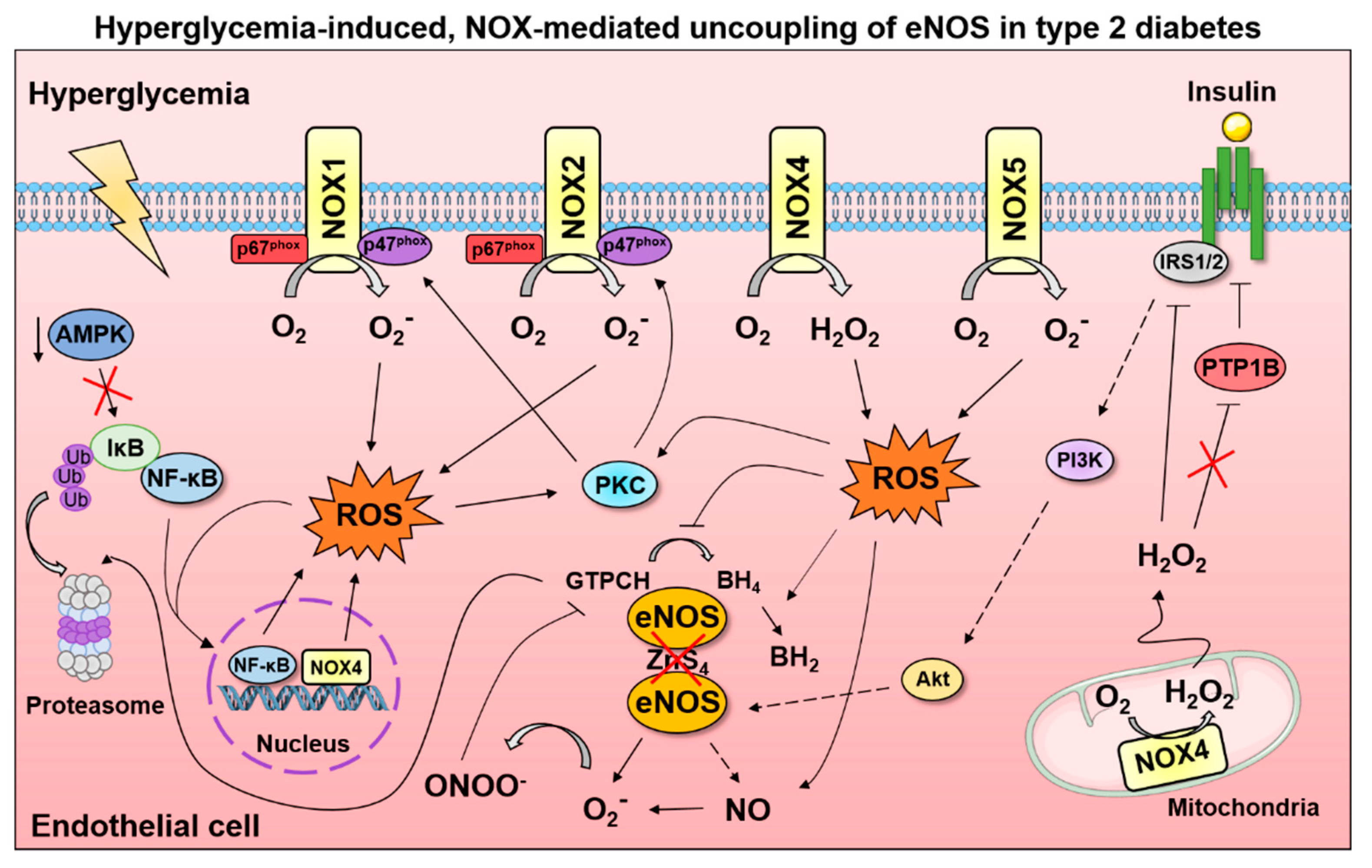{kind=link}
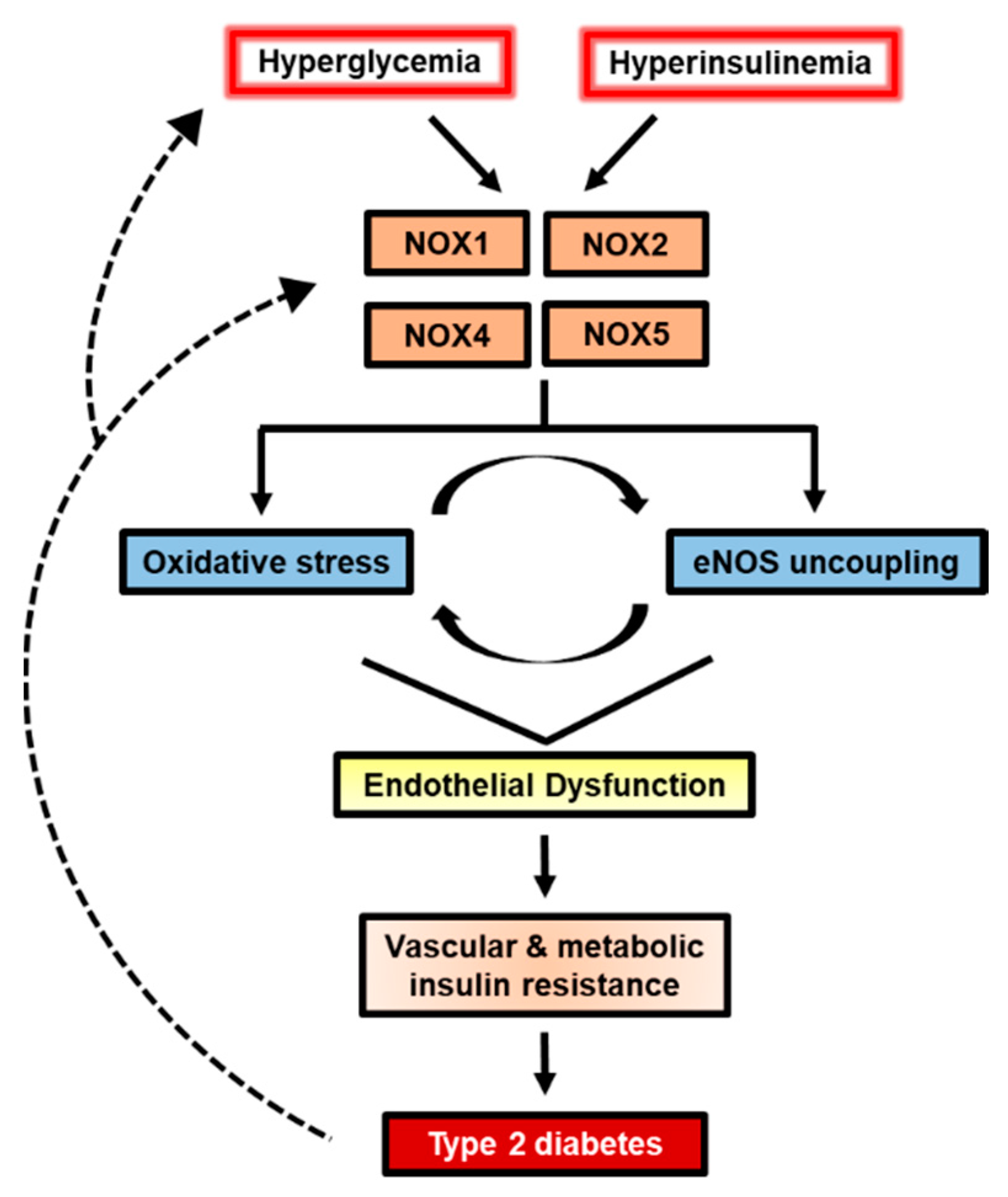{kind=link}
1. Introduction
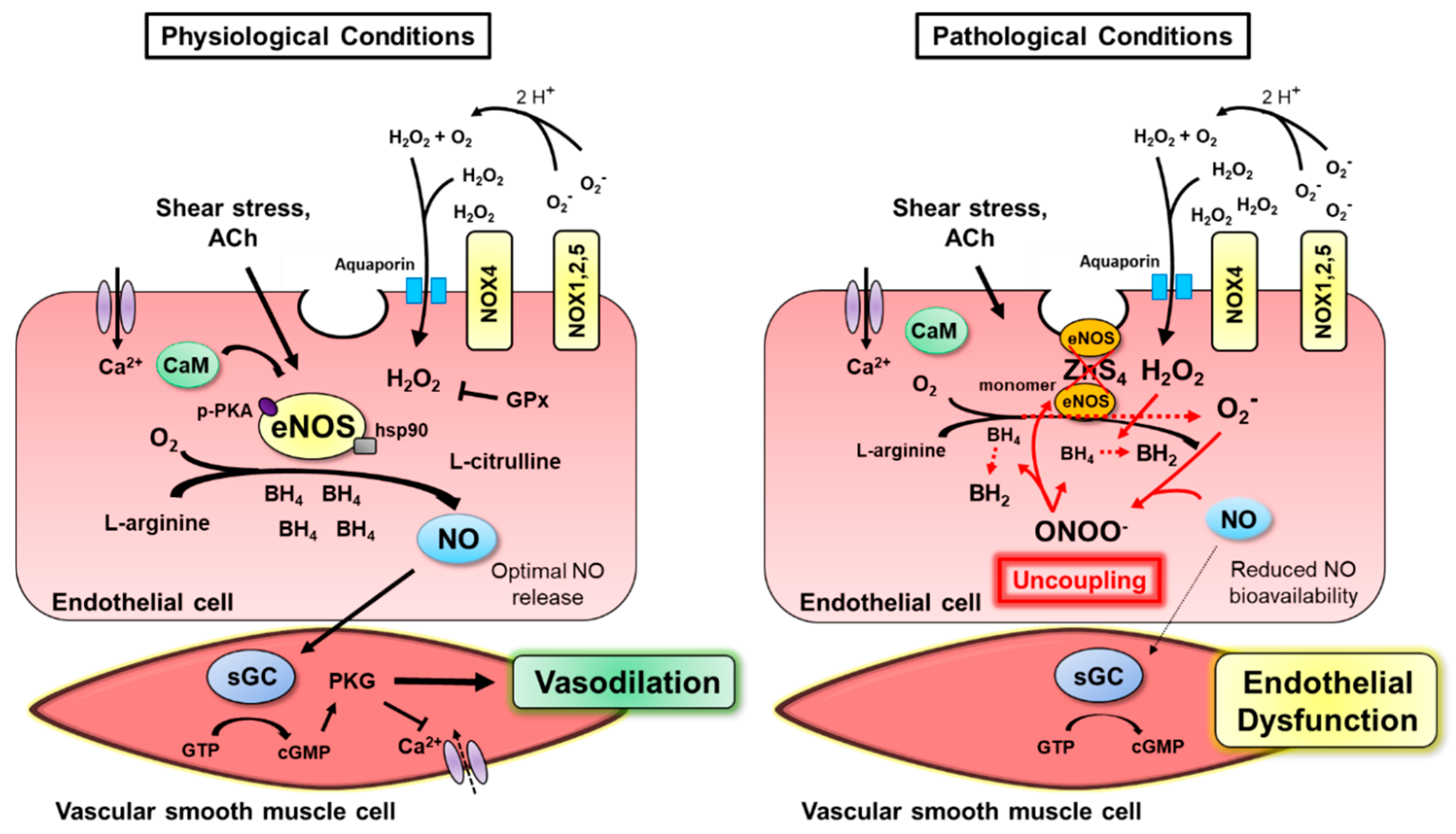
2. Normal Endothelial Function
2.1. NOS Isoforms Mediate Endothelium-Dependent Dilation
2.2. Shear Stress Evokes eNOS Activation
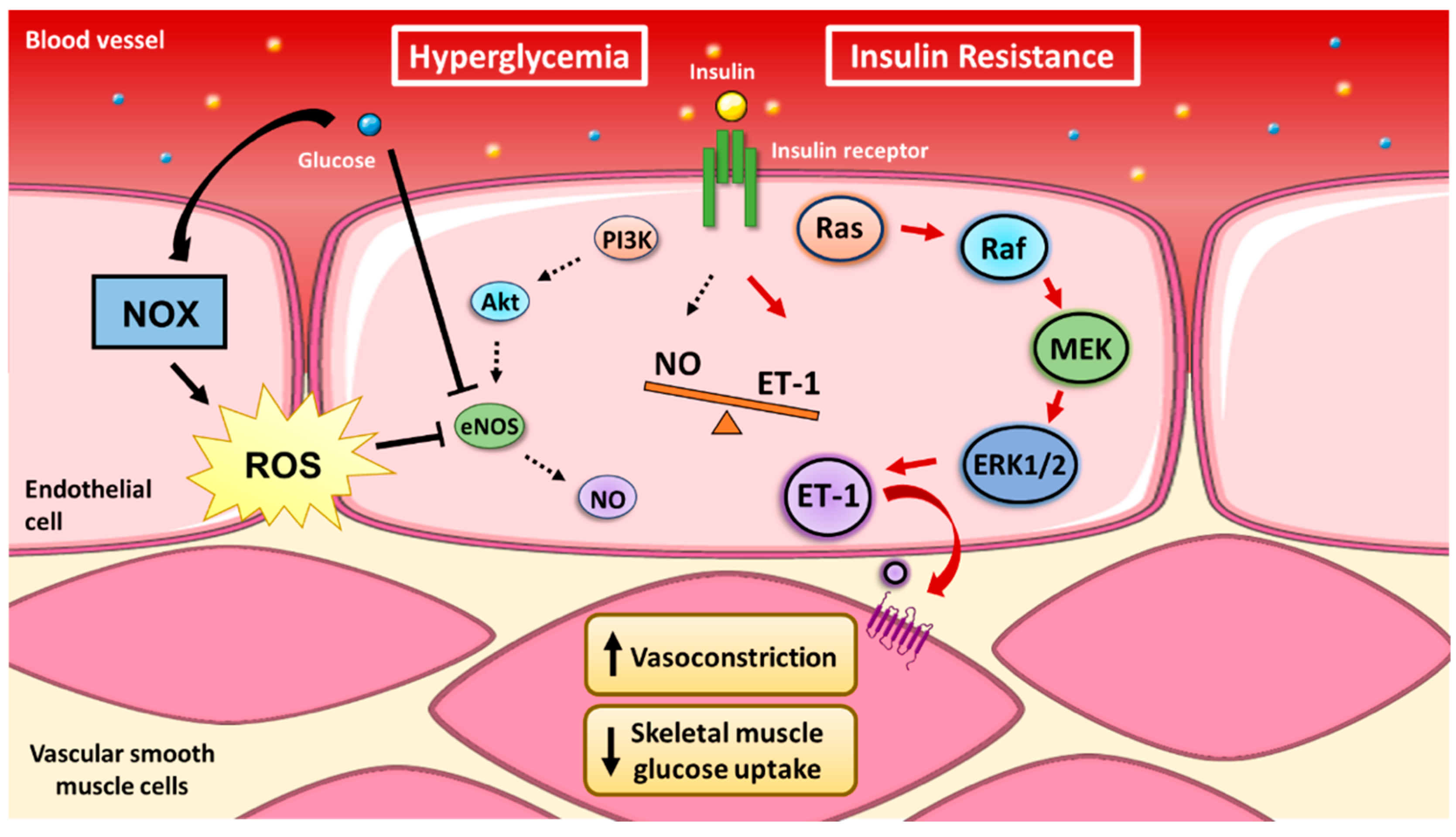
2.3. Coupling of Insulin’s Vasodilatory and Metabolic Functions
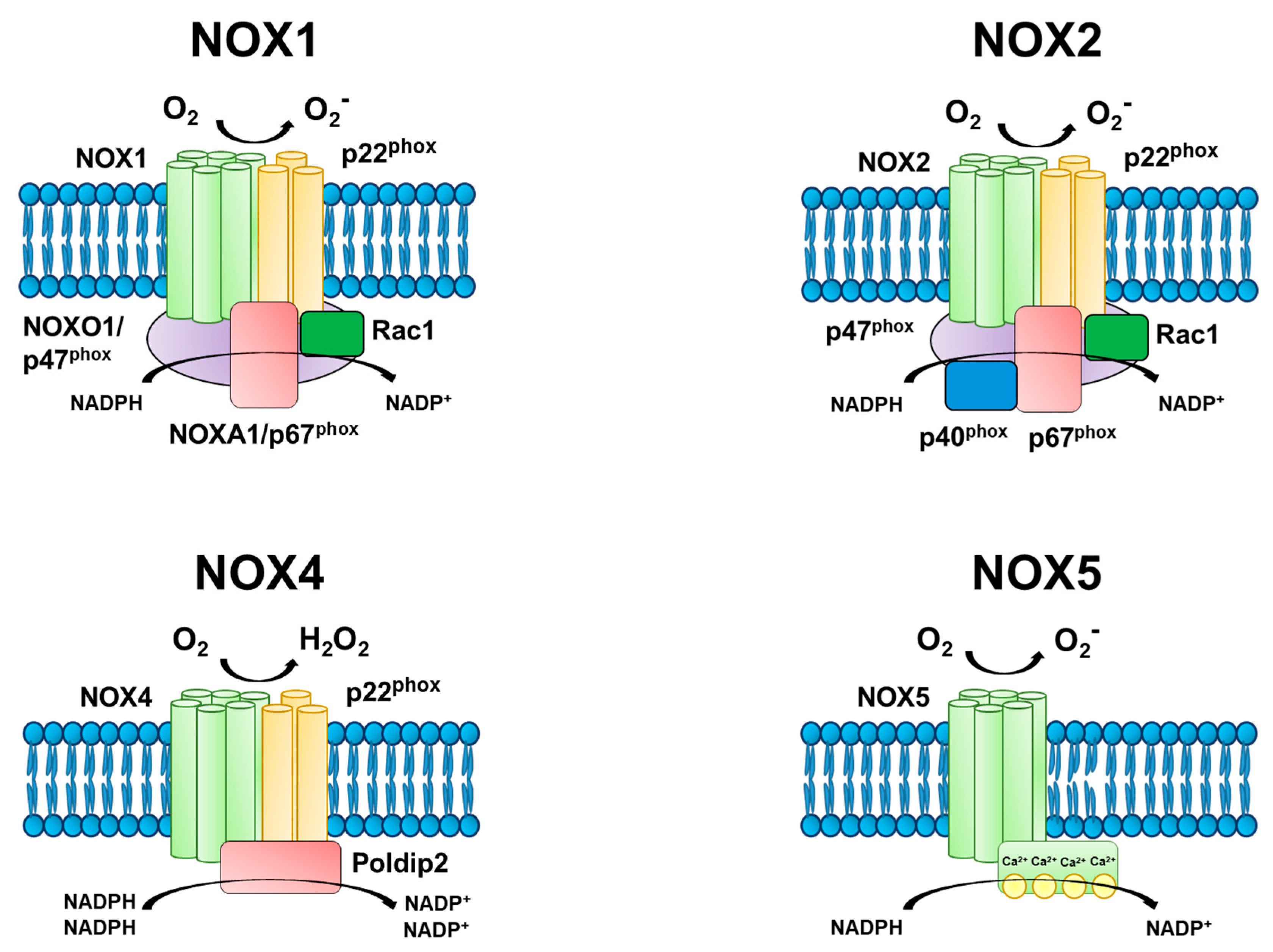
3. Mechanisms of NADPH Oxidase Function
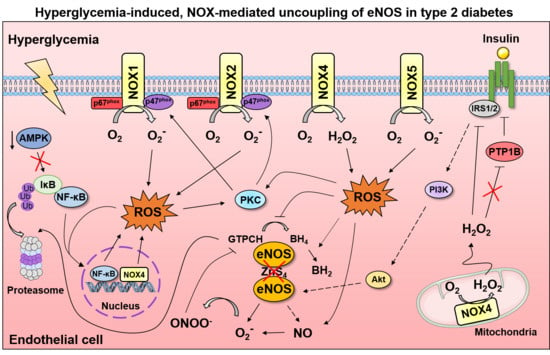
4. Endothelial NOX-derived ROS Production Increases with Exposure to Hyperglycemia
4.1. Endothelial Cell Culture Models of Hyperglycemia
4.2. Endothelial NOX Activity in Animal and Human Clinical Studies
4.2.1. NOX1
4.2.2. NOX2
4.2.3. NOX4
5. Conclusions
Funding
Conflicts of Interest
References
- Wilmot, E.; Idris, I. Early onset type 2 diabetes: Risk factors, clinical impact and management. Adv. Chronic Dis. 2014, 5, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Cardiovascular Disease in Type 2 Diabetes From Population to Man to Mechanisms: The Kelly West Award Lecture 2008. Diabetes Care 2010, 33, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Prigeon, R.L.; McCulloch, D.K.; Boyko, E.J.; Bergman, R.N.; Schwartz, M.W.; Neifing, J.L.; Ward, W.K.; Beard, J.C.; Palmer, J.P. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects: Evidence for a hyperbolic function. Diabetes 1993, 42, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.B.; Goldfine, A.B.; Timimi, F.K.; Ting, H.H.; Roddy, M.-A.; Simonson, D.C.; Creager, M.A. Acute Hyperglycemia Attenuates Endothelium-Dependent Vasodilation in Humans In Vivo. Circulation 1998, 97, 1695–1701. [Google Scholar] [CrossRef]
- Toschi, E.; Camastra, S.; Sironi, A.M.; Masoni, A.; Gastaldelli, A.; Mari, A.; Ferrannini, E.; Natali, A. Effect of Acute Hyperglycemia on Insulin Secretion in Humans. Diabetes 2002, 51, S130–S133. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Rajala, M.W.; Berger, J.P.; Moller, D.E.; Barzilai, N.; Scherer, P.E. Hyperglycemia-induced Production of Acute Phase Reactants in Adipose Tissue. J. Biol. Chem. 2001, 276, 42077–42083. [Google Scholar] [CrossRef]
- Leung, W.K.; Gao, L.; Siu, P.M.; Lai, C.W. Diabetic nephropathy and endothelial dysfunction: Current and future therapies, and emerging of vascular imaging for preclinical renal-kinetic study. Life Sci. 2016, 166, 121–130. [Google Scholar] [CrossRef]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial Dysfunction in Diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34, S285–S290. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Simonson, D.C.; Katz, L.D.; Reichard Jr, G.; Bevilacqua, S.; Barrett, E.J.; Olsson, M.; DeFronzo, R.A. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism 1988, 37, 79–85. [Google Scholar] [CrossRef]
- Walsh, L.K.; Ghiarone, T.; Olver, T.D.; Medina-Hernandez, A.; Edwards, J.C.; Thorne, P.K.; Emter, C.A.; Lindner, J.R.; Manrique-Acevedo, C.; Martinez-Lemus, L.A.; et al. Increased endothelial shear stress improves insulin-stimulated vasodilatation in skeletal muscle. J. Physiol. 2019, 597, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Hatley, M.E.; Bolick, D.T.; Palmer, L.A.; Edelstein, D.; Brownlee, M.; Hedrick, C.C. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 2004, 47, 1727–1734. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. High Glucose Induces Antioxidant Enzymes in Human Endothelial Cells in Culture. Diabetes 1996, 45, 7. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Shen, H.; Liu, H.; Wang, Y.; Bai, Y.; Han, P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zheng, S.; Metreveli, N.S.; Epstein, P.N. Protection of Cardiac Mitochondria by Overexpression of MnSOD Reduces Diabetic Cardiomyopathy. Diabetes 2006, 55, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Minchenko, A.G.; Marinescu, V.; Fathallah, L.; Kennedy, A.; Stockert, C.M.; Frank, R.N.; Stevens, M.J. Antioxidants attenuate early up regulation of retinal vascular endothelial growth factor in streptozotocin-diabetic rats. Diabetologia 2001, 44, 1102–1110. [Google Scholar] [CrossRef]
- Cosentino, F.; Eto, M.; De Paolis, P.; van der Loo, B.; Bachschmid, M.; Ullrich, V.; Kouroedov, A.; Delli Gatti, C.; Joch, H.; Volpe, M.; et al. High Glucose Causes Upregulation of Cyclooxygenase-2 and Alters Prostanoid Profile in Human Endothelial Cells: Role of Protein Kinase C and Reactive Oxygen Species. Circulation 2003, 107, 1017–1023. [Google Scholar] [CrossRef]
- Inoguchi, T. Protein Kinase C-Dependent Increase in Reactive Oxygen Species (ROS) Production in Vascular Tissues of Diabetes: Role of Vascular NAD(P)H Oxidase. J. Am. Soc. Nephrol. 2003, 14, 227–232. [Google Scholar] [CrossRef]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Block, K. Nox as a target for diabetic complications. Clin. Sci. 2013, 125, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Javesghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N.A. Molecular Characterization of a Superoxide-Generating NAD(P)H Oxidase in the Ventilatory Muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef] [PubMed]
- La Favor, J.D.; Dubis, G.S.; Yan, H.; White, J.D.; Nelson, M.A.M.; Anderson, E.J.; Hickner, R.C. Microvascular Endothelial Dysfunction in Sedentary, Obese Humans Is Mediated by NADPH Oxidase: Influence of Exercise Training. Arter. Thromb. Vasc. Biol. 2016, 36, 2412–2420. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadski, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Raman, C.S.; Li, H. Crystal Structure of Constitutive Endothelial Nitric Oxide Synthase: A Paradigm for Pterin Function Involving a Novel Metal Center. Cell 1998, 23, 939–950. [Google Scholar] [CrossRef]
- Daff, S. NO synthase: Structures and mechanisms. Nitric Oxide 2010, 23, 1–11. [Google Scholar] [CrossRef]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef]
- Winger, J.A.; Marletta, M.A. Expression and Characterization of the Catalytic Domains of Soluble Guanylate Cyclase: Interaction with the Heme Domain †. Biochemistry 2005, 44, 4083–4090. [Google Scholar] [CrossRef]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen Reduction by Nitric-oxide Synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef]
- Faraci, F.M.; Didion, S.P. Vascular Protection: Superoxide Dismutase Isoforms in the Vessel Wall. Arter. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Laursen, J.B.; Somers, M.; Kurz, S.; McCann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Endothelial Regulation of Vasomotion in ApoE-Deficient Mice: Implications for Interactions Between Peroxynitrite and Tetrahydrobiopterin. Circulation 2001, 103, 1282–1288. [Google Scholar] [CrossRef]
- Castro, L.; Rodriguez, M.; Radif, R. Aconitase Is Readily Inactivated by Peroxynitrite, but Not by Its Precursor, Nitric Oxide. J. Biol. Chem. 1994, 269, 29409–29415. [Google Scholar]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Alp, N.J.; McAteer, M.A.; Khoo, J.; Choudhury, R.P.; Channon, K.M. Increased Endothelial Tetrahydrobiopterin Synthesis by Targeted Transgenic GTP-Cyclohydrolase I Overexpression Reduces Endothelial Dysfunction and Atherosclerosis in ApoE-Knockout Mice. Arter. Thromb. Vasc. Biol. 2004, 24, 445–450. [Google Scholar] [CrossRef]
- Chen, W.; Druhan, L.J.; Chen, C.-A.; Hemann, C.; Chen, Y.-R.; Berka, V.; Tsai, A.-L.; Zweier, J.L. Peroxynitrite Induces Destruction of the Tetrahydrobiopterin and Heme in Endothelial Nitric Oxide Synthase: Transition from Reversible to Irreversible Enzyme Inhibition. Biochemistry 2010, 49, 3129–3137. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of vascular nitric oxide and reactive oxygen secies and their regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Cho, H.J.; Xie, Q.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Nathan, C. Calmodulin Is a Subunit of Nitric Oxide Synthase from Macrophages. J. Exp. Med. 1992, 176, 599–604. [Google Scholar] [CrossRef]
- Miyoshi, T.; Li, Y.; Shih, D.M.; Wang, X.; Laubach, V.E.; Matsumoto, A.H.; Helm, G.A.; Lusis, A.J.; Shi, W. Deficiency of inducible NO synthase reduces advanced but not early atherosclerosis in apolipoprotein E-deficient mice. Life Sci. 2006, 79, 525–531. [Google Scholar] [CrossRef]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef]
- Fujimoto, M.; Shimizu, N.; Kunii, K.; Martyn, J.A.J.; Ueki, K.; Kaneki, M. A Role for iNOS in Fasting Hyperglycemia and Impaired Insulin Signaling in the Liver of Obese Diabetic Mice. Diabetes 2005, 54, 1340–1348. [Google Scholar] [CrossRef]
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143. [Google Scholar] [CrossRef]
- Kraus, R.M.; Houmard, J.A.; Kraus, W.E.; Tanner, C.J.; Pierce, J.R.; Choi, M.D.; Hickner, R.C. Obesity, insulin resistance, and skeletal muscle nitric oxide synthase. J. Appl. Physiol. 2012, 113, 758–765. [Google Scholar] [CrossRef]
- Huang, A.; Sun, D.; Shesely, E.G.; Levee, E.M.; Koller, A.; Kaley, G. Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am. J. Physiol.-Heart Circ. Physiol. 2002, 282, H429–H436. [Google Scholar] [CrossRef]
- Capettini, L.; Cortes, S.; Silva, J.; Alvarez-Leite, J.; Lemos, V. Decreased production of neuronal NOS-derived hydrogen peroxide contributes to endothelial dysfunction in atherosclerosis. Br. J. Pharm. 2011, 164, 1738–1748. [Google Scholar] [CrossRef]
- Silvagno, F.; Xia, H.; Bredt, D.S. Neuronal Nitric-oxide Synthase-, an Alternatively Spliced Isoform Expressed in Differentiated Skeletal Muscle. J. Biol. Chem. 1996, 271, 11204–11208. [Google Scholar] [CrossRef]
- Baum, O.; Schläppi, S.; Huber-Abel, F.A.; Weichert, A.; Hoppeler, H.; Zakrzewicz, A. The beta-isoform of neuronal nitric oxide synthase (nNOS) lacking the PDZ domain is localized at the sarcolemma. Febs Lett. 2011, 585, 3219–3223. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.; Lin, X.; McConell, G.K. Normal increases in insulin-stimulated glucose uptake after ex vivo contraction in neuronal nitric oxide synthase mu (nNOSμ) knockout mice.pdf. Eur. J. Physiol. 2019, 471, 961–969. [Google Scholar] [CrossRef]
- Putzke, J.; Seidel, B.; Huang, P.L.; Wolf, G. Differential expression of alternatively spliced isoforms of neuronal nitric oxide synthase (nNOS) and N-methyl-D-aspartate receptors (NMDAR) in knockout mice deficient in nNOSa (nNOSaD /D mice). Mol. Brain Res. 2000, 11. [Google Scholar] [CrossRef]
- Bradley, S.J.; Kingwell, B.A.; Canny, B.J.; McConell, G.K. Skeletal muscle neuronal nitric oxide synthase μ protein is reduced in people with impaired glucose homeostasis and is not normalized by exercise training.pdf. Metab.-Clin. Exp. 2007, 56, 1405–1411. [Google Scholar] [CrossRef]
- Hong, Y.H.; Frugier, T.; Zhang, X.; Murphy, R.M.; Lynch, G.S.; Betik, A.C.; Rattigan, S.; McConell, G.K. Glucose uptake during contraction in isolated skeletal muscles from neuronal nitric oxide synthase mu knockout mice. J. Appl. Physiol. 2015, 118, 1113–1121. [Google Scholar] [CrossRef]
- Brandes, R.P.; Kim, D.; Schmitz-Winnenthal, F.-H.; Amidi, M.; Gödecke, A.; Mülsch, A.; Busse, R. Increased Nitrovasodilator Sensitivity in Endothelial Nitric Oxide Synthase Knockout Mice: Role of Soluble Guanylyl Cyclase. Hypertension 2000, 35, 231–236. [Google Scholar] [CrossRef]
- Chatterjee, S. Endothelial Mechanotransduction, Redox Signaling and the Regulation of Vascular Inflammatory Pathways. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Shen, J.; Luscinskas, F.W.; Connolly, A.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Fluid shear stress modulates cytosolic free calcium in vascular endothelial cells. Am. J. Physiol. 1992, 262, C384–C390. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.-S.; Chien, S. Shear Stress–Initiated Signaling and Its Regulation of Endothelial Function. Arter. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Michell, B.J.; Chen, Z.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated Control of Endothelial Nitric-oxide Synthase Phosphorylation by Protein Kinase C and the cAMP-dependent Protein Kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.-P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Boo, Y.C.; Hwang, J.; Sykes, M.; Michell, B.J.; Kemp, B.E.; Lum, H.; Jo, H. Shear stress stimulates phosphorylation of eNOS at Ser 635 by a protein kinase A-dependent mechanism. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H1819–H1828. [Google Scholar] [CrossRef]
- Bauer, P.M.; Fulton, D.; Boo, Y.C.; Sorescu, G.P.; Kemp, B.E.; Jo, H.; Sessa, W.C. Compensatory Phosphorylation and Protein-Protein Interactions Revealed by Loss of Function and Gain of Function Mutants of Multiple Serine Phosphorylation Sites in Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2003, 278, 14841–14849. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Chen, C.-A.; De Pascali, F.; Basye, A.; Hemann, C.; Zweier, J.L. Redox Modulation of Endothelial Nitric Oxide Synthase by Glutaredoxin-1 through Reversible Oxidative Post-Translational Modification. Biochemistry 2013, 52, 6712–6723. [Google Scholar] [CrossRef]
- Baratchi, S.; Khoshmanesh, K.; Woodman, O.L.; Potocnik, S.; Peter, K.; McIntyre, P. Molecular Sensors of Blood Flow in Endothelial Cells. Trends Mol. Med. 2017, 23, 850–868. [Google Scholar] [CrossRef]
- Minshall, R.D.; Sessa, W.C.; Stan, R.V.; Anderson, R.G.W.; Malik, A.B. Caveolin regulation of endothelial function. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 285, L1179–L1183. [Google Scholar]
- Zheng, C.; Liu, Z. Vascular function, insulin action, and exercise: An intricate interplay. Trends Endocrinol. Metab. 2015, 26, 297–304. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Breslin, J.W.; Perrin, R.; Gaudreault, N.; Guo, M.; Kargozaran, H.; Wu, M.H. Microvascular Permeability in Diabetes and Insulin Resistance. Microcirculation 2007, 14, 363–373. [Google Scholar] [CrossRef]
- Vincent, M.A.; Barrett, E.J.; Lindner, J.R.; Clark, M.G.; Rattigan, S. Inhibiting NOS blocks microvascular recruitment and blunts muscle glucose uptake in response to insulin. Am. J. Physiol-Endocrinol. Metab. 2003, 285, E123–E129. [Google Scholar] [CrossRef]
- Vincent, M.A.; Clerk, L.H.; Lindner, J.R.; Klibanov, A.L.; Clark, M.G.; Rattigan, S.; Barrett, E.J. Microvascular Recruitment Is an Early Insulin Effect That Regulates Skeletal Muscle Glucose Uptake In Vivo. Diabetes 2004, 53, 1418–1423. [Google Scholar] [CrossRef]
- Vincent, M.A.; Montagnani, M.; Quon, M.J. Molecular and physiologic actions of insulin related to production of nitric oxide in vascular endothelium. Curr. Diab. Rep. 2003, 3, 279–288. [Google Scholar] [CrossRef]
- Westerbacka, J.; Vehkavaara, S.; Bergholm, R.; Wilkinson, I.; Cockcroft, J.; Yki-Jarvinen, H. Marked resistance of the ability of insulin to decrease arterial stiffness characterizes human obesity. Diabetes 1999, 48, 821–827. [Google Scholar] [CrossRef]
- Baron, A.D. Hemodynamic actions of insulin. Am. J. Physiol.-Endocrinol. Metab. 1994, 267, E187–E202. [Google Scholar] [CrossRef]
- De Filippis, E.; Cusi, K.; Ocampo, G.; Berria, R.; Buck, S.; Consoli, A.; Mandarino, L.J. Exercise-Induced Improvement in Vasodilatory Function Accompanies Increased Insulin Sensitivity in Obesity and Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2006, 91, 4903–4910. [Google Scholar] [CrossRef][Green Version]
- Thijssen, D.H.J.; Dawson, E.A.; Black, M.A.; Hopman, M.T.E.; Cable, N.T.; Green, D.J. Brachial Artery Blood Flow Responses to Different Modalities of Lower Limb Exercise. Med. Sci. Sports Exerc. 2009, 41, 1072–1079. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lin, Y.-W.; Clemont, A.; Feener, E.P.; Hein, K.D.; Igarashi, M.; Yamauchi, T.; White, M.F.; King, G.L. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J. Clin. Investig. 1999, 104, 447–457. [Google Scholar] [CrossRef]
- Kim, J.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction: Molecular and Pathophysiological Mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Investig. 1996, 98, 894–898. [Google Scholar] [CrossRef]
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.-N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles for Insulin Receptor, PI3-Kinase, and Akt in Insulin-Signaling Pathways Related to Production of Nitric Oxide in Human Vascular Endothelial Cells. Circulation 2000, 101, 1539–1545. [Google Scholar] [CrossRef]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase– and MAP kinase–mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef]
- Tomlinson, D.R. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia 1999, 42, 1271–1281. [Google Scholar] [CrossRef]
- Ferri, C.; Piccoli, A.; Laurenti, O.; Cassone, M.R.; Bellini, C.; Properzi, G.; Valesini, G.; De Mattia, G.; Santucci, A. Insulin stimulates endothelin-1 secretion from human endothelial cells and modulates its circulating levels in vivo. J. Clin. Endocrinol. Metab. 1995, 80, 829–835. [Google Scholar]
- Wagner, O.F.; Christ, G.; Wojta, J.; Vierhapper, H.; Parzer, S.; Nowotny, P.J.; Schneider, B.; Waldhausl, W.; Binder, B.R. Polar Secretion of Endothelin-1 by Cultured Endothelial Cells. J. Biol. Chem. 1992, 267, 16066–16068. [Google Scholar]
- Ivey, M.E.; Osman, N.; Little, P.J. Endothelin-1 signalling in vascular smooth muscle: Pathways controlling cellular functions associated with atherosclerosis. Atherosclerosis 2008, 199, 237–247. [Google Scholar] [CrossRef]
- Piatti, P.; Monti, L.D.; Conti, M.; Baruffaldi, L.; Galli, L.; Phan, C.V.; Guazzini, B.; Pontiroli, A.E.; Pozza, G. Hypertrigtyceridemia and Hyperinsulinemia Are Potent Inducers of Endothelin-1 Release in Humans. Diabetes 1996, 45, 6. [Google Scholar] [CrossRef]
- Wu, S. Altered paracrine effect of endothelin in blood vessels of the hyperinsulinemic, insulin resistant obese Zucker rat. Cardiovasc. Res. 2000, 45, 994–1000. [Google Scholar] [CrossRef][Green Version]
- Nishiyama, S.K.; Zhao, J.; Wray, D.W.; Richardson, R.S. Vascular function and endothelin-1: Tipping the balance between vasodilation and vasoconstriction. J. Appl. Physiol. 2017, 122, 354–360. [Google Scholar] [CrossRef]
- Mather, K.J.; Mirzamohammadi, B.; Lteif, A.; Steinberg, H.O.; Baron, A.D. Endothelin Contributes to Basal Vascular Tone and Endothelial Dysfunction in Human Obesity and Type 2 Diabetes. Diabetes 2002, 51, 3517–3523. [Google Scholar] [CrossRef]
- Weil, B.R.; Westby, C.M.; Van Guilder, G.P.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Enhanced endothelin-1 system activity with overweight and obesity. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H689–H695. [Google Scholar] [CrossRef]
- Cardillo, C.; Nambi, S.S.; Kilcoyne, C.M.; Choucair, W.K.; Katz, A.; Quon, M.J.; Panza, J.A. Insulin Stimulates Both Endothelin and Nitric Oxide Activity in the Human Forearm. Circulation 1999, 100, 820–825. [Google Scholar] [CrossRef]
- Barton, M.; Haudenschild, C.C.; d’Uscio, L.V.; Shaw, S.; Munter, K.; Luscher, T.F. Endothelin ETA receptor blockade restores NO-mediated endothelial function and inhibits atherosclerosis in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 14367–14372. [Google Scholar] [CrossRef]
- Bromberg, Y.; Pick, E. Unsaturated fatty acids stimulate NADPH-dependent superoxide production by cell-free system derived from macrophages. Cell. Immunol. 1984, 88, 213–221. [Google Scholar] [CrossRef]
- Segal, A.; Abo, A. The biochemical basis of the NADPH oxidase of phagocytes. Trends Biochem. Sci. 1993, 18, 43–47. [Google Scholar] [CrossRef]
- Jones, S.; O’Donnell, V.; Wood, J.; Broughton, J.; Hughes, E.; Jones, O. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am. J. Physiol.-Heart Circ. Physiol. 1996, 271, H1626–H1634. [Google Scholar]
- Magnani, F.; Nenci, S.; Millana Fananas, E.; Ceccon, M.; Romero, E.; Fraaije, M.W.; Mattevi, A. Crystal structures and atomic model of NADPH oxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 6764–6769. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Tamma, G.; Valenti, G.; Grossini, E.; Donnini, S.; Marino, A.; Marinelli, R.A.; Calamita, G. Aquaporin Membrane Channels in Oxidative Stress, Cell Signaling, and Aging: Recent Advances and Research Trends. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Touyz, R.M. NADPH Oxidases, Reactive Oxygen Species, and Hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31, S170–S180. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.-H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop Is Involved in Hydrogen Peroxide Formation by the NADPH Oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Dikalov, S.; Dikalova, A.; Bikineyeva, A.; Schmidt, H.; Harrison, D.; Griendling, K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A Hydrogen Peroxide-Generating Oxygen Sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef]
- Buul, J.D.V.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxid. Redox Signal. 2005, 7, 308–317. [Google Scholar] [CrossRef]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct Subcellular Localizations of Nox1 and Nox4 in Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef]
- Clark, R.A.; Valente, A.J. Nuclear factor kappa B activation by NADPH oxidases. Mech. Ageing Dev. 2004, 125, 799–810. [Google Scholar] [CrossRef]
- Sullivan-Gunn, M.J.; Lewandowski, P.A. Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr. 2013, 13. [Google Scholar] [CrossRef]
- Park, K.; Gross, M.; Lee, D.-H.; Holvoet, P.; Himes, J.H.; Shikany, J.M.; Jacobs, D.R. Oxidative Stress and Insulin Resistance: The Coronary Artery Risk Development in Young Adults study. Diabetes Care 2009, 32, 1302–1307. [Google Scholar] [CrossRef]
- Taye, A.; Saad, A.H.; Kumar, A.H.; Morawietz, H. Effect of apocynin on NADPH oxidase-mediated oxidative stress-LOX-1-eNOS pathway in human endothelial cells exposed to high glucose. Eur. J. Pharm. 2010, 627, 42–48. [Google Scholar] [CrossRef]
- Ding, H.; Aljofan, M.; Triggle, C.R. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J. Cell. Physiol. 2007, 212, 682–689. [Google Scholar] [CrossRef]
- Adela, R.; Nethi, S.K.; Bagul, P.K.; Barui, A.K.; Mattapally, S.; Kuncha, M.; Patra, C.R.; Reddy, P.N.C.; Banerjee, S.K. Hyperglycaemia Enhances Nitric Oxide Production in Diabetes: A Study from South Indian Patients. PLoS ONE 2015, 10, e0125270. [Google Scholar] [CrossRef]
- Noronha, B.T.; Li, J.-M.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.T. Inducible Nitric Oxide Synthase Has Divergent Effects on Vascular and Metabolic Function in Obesity. Diabetes 2005, 54, 1082–1089. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial Nitric Oxide Synthase Uncoupling Impairs Endothelial Progenitor Cell Mobilization and Function in Diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.-H. Proteasome-Dependent Degradation of Guanosine 5′-Triphosphate Cyclohydrolase I Causes Tetrahydrobiopterin Deficiency in Diabetes Mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Zhu, H.; Song, P.; Zou, M.-H. Peroxynitrite-Dependent Zinc Release and Inactivation of Guanosine 5’-Triphosphate Cyclohydrolase 1 Instigate Its Ubiquitination in Diabetes. Diabetes 2013, 62, 4247–4256. [Google Scholar] [CrossRef]
- Zhang, Q.; Malik, P.; Pandey, D.; Gupta, S.; Jagnandan, D.; de Chantemele, E.B.; Banfi, B.; Marrero, M.B.; Rudic, R.D.; Stepp, D.W.; et al. Paradoxical Activation of Endothelial Nitric Oxide Synthase by NADPH Oxidase. Arter. Thromb. Vasc. Biol. 2008, 28, 1627–1633. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Wauquier, F.; Eid, A.A.; Roman, L.J.; Ghosh-Choudhury, G.; Khazim, K.; Block, K.; Gorin, Y. Nox4 NADPH Oxidase Mediates Peroxynitrite-dependent Uncoupling of Endothelial Nitric-oxide Synthase and Fibronectin Expression in Response to Angiotensin II: Role of Mitochondrial Reactive Oxygen Species. J. Biol. Chem. 2013, 288, 28668–28686. [Google Scholar] [CrossRef]
- Chen, F.; Qian, L.-H.; Deng, B.; Liu, Z.-M.; Zhao, Y.; Le, Y.-Y. Resveratrol Protects Vascular Endothelial Cells from High Glucose-Induced Apoptosis through Inhibition of NADPH Oxidase Activation-Driven Oxidative Stress. CNS Neurosci. Ther. 2013, 19, 675–681. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, Inflammation, and Metabolic Disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef]
- Williams, C.R.; Lu, X.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates NF-kB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C213–C223. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St.-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting cAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Chao, L.; Marcus-Samuels, B.; Mason, M.M.; Moitra, J.; Vinson, C.; Arioglu, E.; Gavrilova, O.; Reitman, M.L. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J. Clin. Investig. 2000, 106, 1221–1228. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone Reduces Glucose-Induced Oxidative Stress Mediated by NAD(P)H Oxidase via AMPK-Dependent Mechanism. Arter.Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell Signaling through Protein Kinase C Oxidation and Activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef]
- Rahman, A.; Anwar, K.N.; Malik, A.B. Protein kinase C-ζ mediates TNF-α-induced ICAM-1 gene transcription in endothelial cells.pdf. Am. J. Physiol. Cell Physiol. 2000, 279, C906–C914. [Google Scholar] [CrossRef]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-βI and prooxidant enzyme NADPH oxidase. Redox. Biol. 2014, 2, 694–701. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of Increased Vascular Superoxide Production in Human Diabetes Mellitus: Role of NAD(P)H Oxidase and Endothelial Nitric Oxide Synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef]
- Tailor, I.; Tennenbaum, T.; Kuroki, T.; Eldar-Finkelman, H. PKC-δ-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice_ role for NADPH oxidase. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E405–E411. [Google Scholar] [CrossRef][Green Version]
- Hirst, J.A.; Farmer, A.J.; Ali, R.; Roberts, N.W.; Stevens, R.J. Quantifying the Effect of Metformin Treatment and Dose on Glycemic Control. Diabetes Care 2012, 35, 446–454. [Google Scholar] [CrossRef]
- Gallo, A.; Ceolotto, G.; Pinton, P.; Iori, E.; Murphy, E.; Rutter, G.A.; Rizzuto, R.; Semplicini, A.; Avogaro, A. Metformin Prevents Glucose-Induced Protein Kinase C- 2 Activation in Human Umbilical Vein Endothelial Cells Through an Antioxidant Mechanism. Diabetes 2005, 54, 1123–1131. [Google Scholar] [CrossRef][Green Version]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells: The Role of Protein Kinase C and NAD(P)H-Oxidase Activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, M.; Liang, B.; Xu, J.; Xie, Z.; Chao, L.; Benoit, V.; Daoguang, Y.; Zou, M.-J. AMPKα2 Deletion Causes Aberrant Expression and Activation of NAD(P)H Oxidase and Consequent Endothelial Dysfunction In Vivo. Circ. Res. 2010, 106, 1117–1128. [Google Scholar] [CrossRef]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.K.; Savinko, T.; Wong, A.K.F.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef]
- Breuss, J.; Atanasov, A.; Uhrin, P. Resveratrol and Its Effects on the Vascular System. Int. J. Mol. Sci. 2019, 20, 1523. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Rosiglitazone Revisited: An Updated Meta-analysis of Risk for Myocardial Infarction and Cardiovascular Mortality. Arch. Intern. Med. 2010, 170. [Google Scholar] [CrossRef]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating Glucose Is More Deleterious to Endothelial Function and Oxidative Stress Than Mean Glucose in Normal and Type 2 Diabetic Patients. Diabetes 2008, 57, 1349–1354. [Google Scholar] [CrossRef]
- An, H.; Wei, R.; Ke, J.; Yang, J.; Liu, Y.; Wang, X.; Wang, G.; Hong, T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J. Diabetes Complicat. 2016, 30, 1017–1024. [Google Scholar] [CrossRef]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent High Glucose Implements Stress-Induced Senescence in Human Vascular Endothelial Cells: Role of Superoxide Production by NADPH Oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Stefano, G.B.; Challenger, S.; Kream, R.M. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur. J. Nutr. 2016, 55, 2339–2345. [Google Scholar] [CrossRef]
- Sada, K.; Nishikawa, T.; Kukidome, D.; Yoshinaga, T.; Kajihara, N.; Sonoda, K.; Senokuchi, T.; Motoshima, H.; Matsumura, T.; Araki, E. Hyperglycemia Induces Cellular Hypoxia through Production of Mitochondrial ROS Followed by Suppression of Aquaporin-1. PLoS ONE 2016, 11, e0158619. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-Activated Protein Kinase Reduces Hyperglycemia-Induced Mitochondrial Reactive Oxygen Species Production and Promotes Mitochondrial Biogenesis in Human Umbilical Vein Endothelial cells. Diabetes 2006, 55, 8. [Google Scholar] [CrossRef]
- Dymkowska, D.; Drabarek, B.; Podszywałow-Bartnicka, P.; Szczepanowska, J.; Zabłocki, K. Hyperglycaemia modifies energy metabolism and reactive oxygen species formation in endothelial cells in vitro. Arch. Biochem. Biophys. 2014, 542, 7–13. [Google Scholar] [CrossRef]
- Graham, N.A.; Tahmasian, M.; Kohli, B.; Komisopoulou, E.; Zhu, M.; Vivanco, I.; Teitell, M.A.; Wu, H.; Ribas, A.; Lo, R.S.; et al. Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol. Syst. Biol. 2012, 8. [Google Scholar] [CrossRef]
- BelAiba, R.S.; Djordjevic, T.; Petry, A.; Diemer, K.; Bonello, S.; Banfi, B.; Hess, J.; Pogrebniak, A.; Bickel, C.; Gorlach, A. NOX5 variants are functionally active in endothelial cells. Free Radic. Biol. Med. 2007, 42, 446–459. [Google Scholar] [CrossRef]
- Jay, D.B.; Papaharalambus, C.A.; Seidel-Rogol, B.; Dikalova, A.E.; Lassègue, B.; Griendling, K.K. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 329–335. [Google Scholar] [CrossRef]
- Hahn, N.E.; Meischl, C.; Kawahara, T.; Musters, R.J.P.; Verhoef, V.M.J.; van der Velden, J.; Vonk, A.B.A.; Paulus, W.J.; van Rossum, A.C.; Niessen, H.W.M.; et al. NOX5 Expression Is Increased in Intramyocardial Blood Vessels and Cardiomyocytes after Acute Myocardial Infarction in Humans. Am. J. Pathol. 2012, 180, 2222–2229. [Google Scholar] [CrossRef]
- Chen, F.; Wang, Y.; Barman, S.; Fulton, D. Enzymatic regulation and functional relevance of NOX5. Curr. Pharm. Des. 2015, 21, 5999–6008. [Google Scholar] [CrossRef]
- Pandey, D.; Patel, A.; Patel, V.; Chen, F.; Qian, J.; Wang, Y.; Barman, S.A.; Venema, R.C.; Stepp, D.W.; Daniel Rudic, R.; et al. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H1919–H1928. [Google Scholar] [CrossRef]
- Jha, J.C.; Watson, A.M.D.; Mathew, G.; de Vos, L.C.; Jandeleit-Dahm, K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin. Sci. 2017, 131, 981–990. [Google Scholar] [CrossRef]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019. [Google Scholar] [CrossRef]
- Chen, F.; Yu, Y.; Haigh, S.; Johnson, J.; Lucas, R.; Stepp, D.W.; Fulton, D.J.R. Regulation of NADPH Oxidase 5 by Protein Kinase C Isoforms. PLoS ONE 2014, 9, e88405. [Google Scholar] [CrossRef]
- Wendt, M.; Daiber, A.; Kleschyov, A.; Mulsch, A.; Sydow, K.; Schulz, E.; Chen, K.; Keaney, J., Jr.; Lassègue, B.; Walter, U.; et al. Differential effects of diabetes on the expression of the gp91phox homologues nox1 and nox4. Free Radic. Biol. Med. 2005, 39, 381–391. [Google Scholar] [CrossRef]
- Youn, J.Y.; Gao, L.; Cai, H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012, 55, 2069–2079. [Google Scholar] [CrossRef]
- Rezende, F.; Moll, F.; Walter, M.; Helfinger, V.; Hahner, F.; Janetzko, P.; Ringel, C.; Weigert, A.; Fleming, I.; Weissmann, N.; et al. The NADPH organizers NoxO1 and p47phox are both mediators of diabetes-induced vascular dysfunction in mice. Redox Biol. 2018, 15, 12–21. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus–Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Hwang, J.; Kleinhenz, D.J.; Rupnow, H.L.; Campbell, A.G.; Thulé, P.M.; Sutliff, R.L.; Hart, C.M. The PPARγ ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vasc. Pharm. 2007, 46, 456–462. [Google Scholar] [CrossRef]
- Chew, P.; Yuen, D.Y.C.; Koh, P.; Stefanovic, N.; Febbraio, M.A.; Kola, I.; Cooper, M.E.; de Haan, J.B. Site-Specific Antiatherogenic Effect of the Antioxidant Ebselen in the Diabetic Apolipoprotein E–Deficient Mouse. Arter. Thromb. Vasc. Biol. 2009, 29, 823–830. [Google Scholar] [CrossRef]
- Ding, H.; Hashem, M.; Triggle, C. Increased oxidative stress in the streptozotocin-induced diabetic apoE-deficient mouse: Changes in expression of NADPH oxidase subunits and eNOS. Eur. J. Pharm. 2007, 561, 121–128. [Google Scholar] [CrossRef]
- Lynch, C.M.; Kinzenbaw, D.A.; Chen, X.; Zhan, S.; Mezzetti, E.; Filosa, J.; Ergul, A.; Faulkner, J.L.; Faraci, F.M.; Didion, S.P. Nox2-Derived Superoxide Contributes to Cerebral Vascular Dysfunction in Diet-Induced Obesity. Stroke 2013, 44, 3195–3201. [Google Scholar] [CrossRef]
- Du, J.; Fan, L.M.; Mai, A.; Li, J.-M. Crucial roles of Nox2-derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle-aged mice: Dietary obesity and vascular oxidative stress. Br. J. Pharm. 2013, 170, 1064–1077. [Google Scholar] [CrossRef]
- Sukumar, P.; Viswambharan, H.; Imrie, H.; Cubbon, R.M.; Yuldasheva, N.; Gage, M.; Galloway, S.; Skromna, A.; Kandavelu, P.; Santos, C.X.; et al. Nox2 NADPH Oxidase Has a Critical Role in Insulin Resistance-Related Endothelial Cell Dysfunction. Diabetes 2013, 62, 2130–2134. [Google Scholar] [CrossRef]
- Di Marco, E.; Gray, S.P.; Chew, P.; Kennedy, K.; Cooper, M.E.; Schmidt, H.H.H.W.; Jandeleit-Dahm, K.A.M. Differential effects of NOX4 and NOX1 on immune cell-mediated inflammation in the aortic sinus of diabetic ApoE-/- mice. Clin. Sci. 2016, 130, 1363–1374. [Google Scholar] [CrossRef]
- Halban, P.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-cell failure in type 2 diabetes_postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W. Vascular Inflammation, Insulin Resistance, and Reduced Nitric Oxide Production Precede the Onset of Peripheral Insulin Resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef]
- Souto Padron de Figueiredo, A.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 Mediates Skeletal Muscle Insulin Resistance Induced by a High Fat Diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef]
- Coats, B.R.; Schoenfelt, K.Q.; Barbosa-Lorenzi, V.C.; Peris, E.; Cui, C.; Hoffman, A.; Zhou, G.; Fernandez, S.; Zhai, L.; Hall, B.A.; et al. Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep. 2017, 20, 3149–3161. [Google Scholar] [CrossRef]
- Pollack, R.M.; Donath, M.Y.; LeRoith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39, S244–S252. [Google Scholar] [CrossRef]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.K.; Warnholtz, A.; et al. Mechanisms Underlying Endothelial Dysfunction in Diabetes Mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Jacob, S.; Kinnick, T.R.; Teachey, M.K.; Krekler, M. Selective Angiotensin II Receptor Antagonism Reduces Insulin Resistance in Obese Zucker Rats. Hypertension 2001, 38, 884–890. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Szczurek, M.R.; Blackburn, B.K.; Mey, J.T.; Chen, Z.; Robinson, A.T.; Bian, J.-T.; Unterman, T.G.; Minshall, R.D.; Brown, M.D.; et al. Hyperinsulinemia augments endothelin-1 protein expression and impairs vasodilation of human skeletal muscle arterioles. Physiol. Rep. 2016, 4, e12895. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ali, M.M.; Miranda, E.R.; Mey, J.T.; Blackburn, B.K.; Haus, J.M.; Phillips, S.A. Nox2 contributes to hyperinsulinemia-induced redox imbalance and impaired vascular function. Redox Biol. 2017, 13, 288–300. [Google Scholar] [CrossRef]
- Espinosa, A.; García, A.; Härtel, S.; Hidalgo, C.; Jaimovich, E. NADPH Oxidase and Hydrogen Peroxide Mediate Insulin-induced Calcium Increase in Skeletal Muscle Cells. J. Biol. Chem. 2009, 284, 2568–2575. [Google Scholar] [CrossRef]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the Major Catalytic Component of an Endothelial NAD(P)H Oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F. NADPH Oxidase 4 Promotes Endothelial Angiogenesis Through Endothelial Nitric Oxide Synthase Activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H Oxidase Homolog Nox4 Modulates Insulin-Stimulated Generation of H2O2 and Plays an Integral Role in Insulin Signal Transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Mahadev, K.; Wu, X.; Zhu, L.; Motoshima, H. Role of Insulin-Induced Reactive Oxygen Species in the Insulin Signaling Pathway. Antioxid. Redox Signal. 2005, 7, 1021–1031. [Google Scholar] [CrossRef]
- Li, Y.; Mouche, S.; Sajic, T.; Veyrat-Durebex, C.; Supale, R.; Pierroz, D.; Ferrari, S.; Negro, F.; Hasler, U.; Feraille, E.; et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int. J. Obes. 2012, 36, 1503–1513. [Google Scholar] [CrossRef]
- Giaccari, A.; Morviducci, L.; Pastore, L.; Zorretta, D.; Sbraccia, P.; Maroccia, E.; Buongiorno, A.; Tamburrano, G. Relative contribution of glycogenolysis and gluconeogenesis to hepatic glucose production in control and diabetic rats. A re-examination in the presence of euglycaemia. Diabetologia 1998, 41, 307–314. [Google Scholar] [CrossRef][Green Version]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.L.; Touyz, R.M.; Cooper, M.E.; et al. Reactive Oxygen Species Can Provide Atheroprotection via NOX4-Dependent Inhibition of Inflammation and Vascular Remodeling. Arter. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arter. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef]
- Antonopoulos, A.S.; Margaritis, M.; Coutinho, P.; Shirodaria, C.; Psarros, C.; Herdman, L.; Sanna, F.; De Silva, R.; Petrou, M.; Sayeed, R.; et al. Adiponectin as a Link Between Type 2 Diabetes and Vascular NADPH Oxidase Activity in the Human Arterial Wall: The Regulatory Role of Perivascular Adipose Tissue. Diabetes 2015, 64, 2207–2219. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Patti, M.-E.; Corvera, S. The Role of Mitochondria in the Pathogenesis of Type 2 Diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.-T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- La Favor, J.D.; Anderson, E.J.; Hickner, R.C. Novel Method for Detection of Reactive Oxygen Species In Vivo in Human Skeletal Muscle. Physiol. Res. 2014, 63, 387–392. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meza, C.A.; La Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. https://doi.org/10.3390/ijms20153775
Meza CA, La Favor JD, Kim D-H, Hickner RC. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? International Journal of Molecular Sciences. 2019; 20(15):3775. https://doi.org/10.3390/ijms20153775
Chicago/Turabian StyleMeza, Cesar A., Justin D. La Favor, Do-Houn Kim, and Robert C. Hickner. 2019. "Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS?" International Journal of Molecular Sciences 20, no. 15: 3775. https://doi.org/10.3390/ijms20153775
APA StyleMeza, C. A., La Favor, J. D., Kim, D.-H., & Hickner, R. C. (2019). Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? International Journal of Molecular Sciences, 20(15), 3775. https://doi.org/10.3390/ijms20153775