Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications



Abstract
1. Introduction
1.1. All-Atom Molecular Dynamics Simulations
1.2. Enhanced Sampling Methods: An Effort to Understand the Larger Picture of Biological Processes
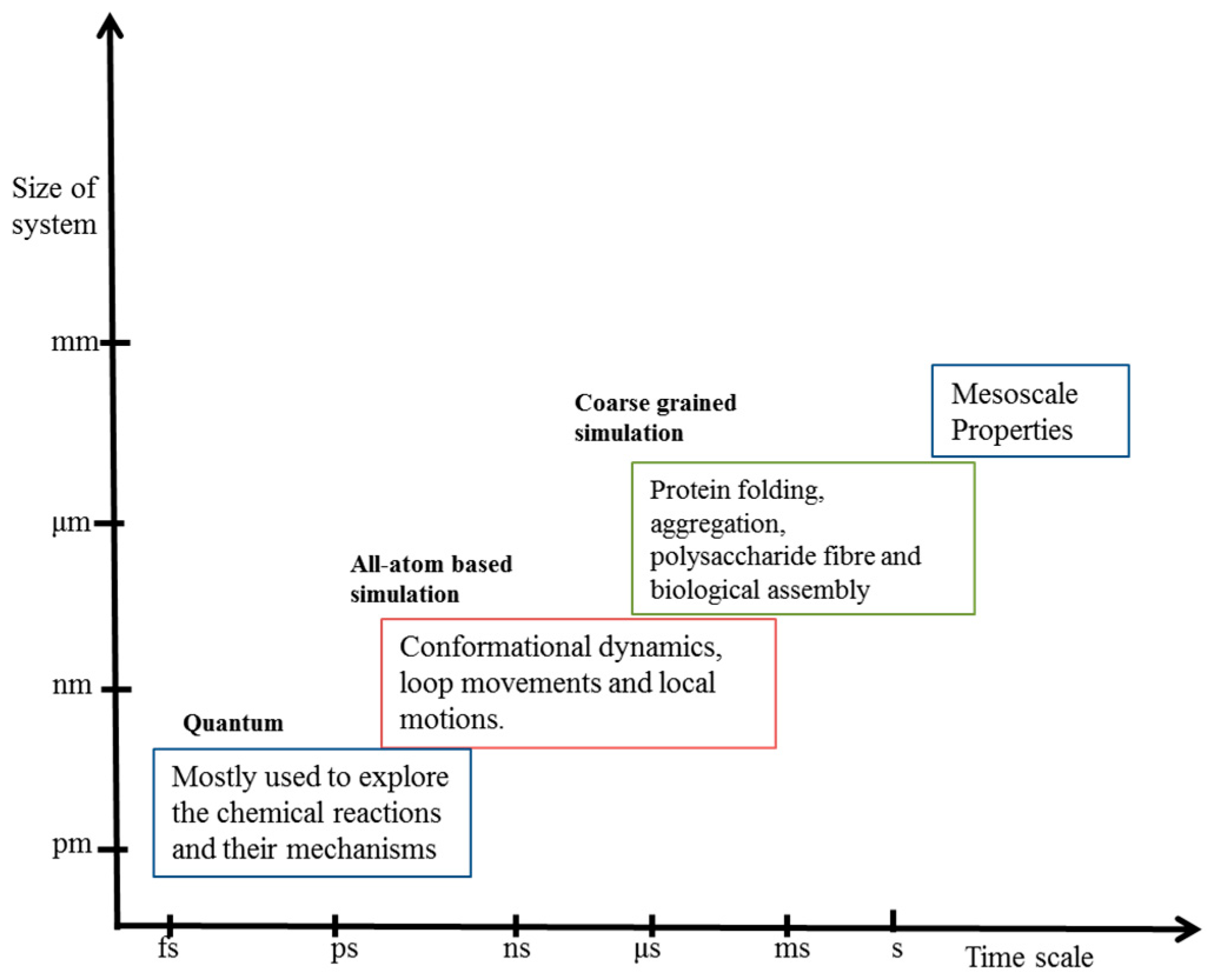
1.3. Coarse-Grained Modeling and Necessity
2. Coarse-Grained Simulation: Basic Principle
2.1. Coarse-Grained Models for Proteins
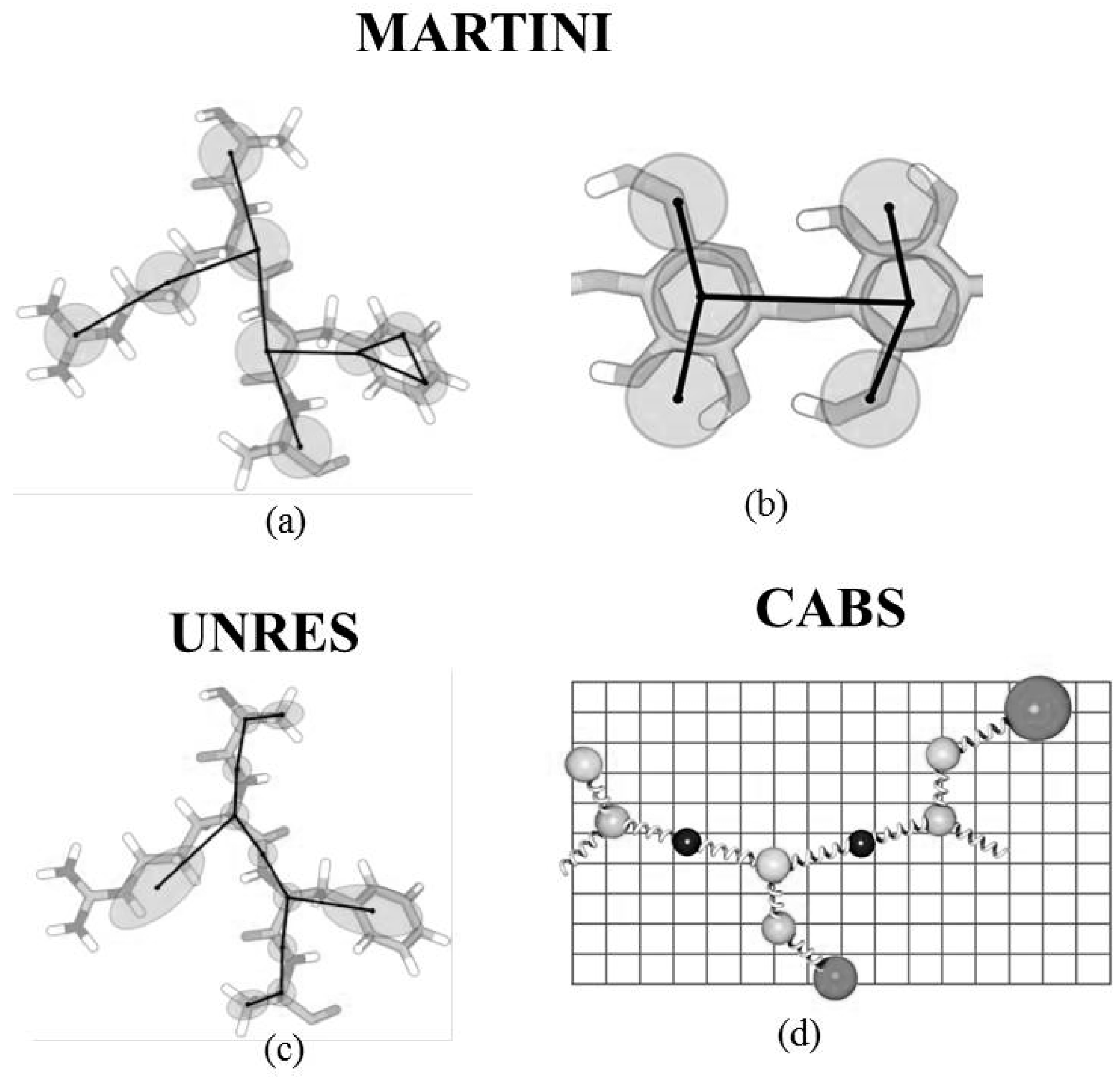
2.1.1. MARTINI Model
2.1.2. UNRES Model
2.1.3. CABS (C-Alpha, Beta and Side Chain)
2.1.4. PRIMO (Protein Intermediate Model)
2.1.5. OPEP (Optimized Potential for Efficient Structure Prediction)
2.1.6. PRIME
2.1.7. Bereau and Deserno Model
2.1.8. Kim and Hummer Model
2.2. Coarse-Grained Models for Carbohydrates
2.2.1. M3B Model
2.2.2. MARTINI Model
2.2.3. Other Models
3. Coarse-Grained Models and Biomolecular Complexes
4. Multiscale Simulations and Coarse-Grained Models
5. Challenges in Coarse-Grained Modeling
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levitt, M. Birth and future of multiscale modeling for macromolecular systems (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2014, 53, 10006–10018. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Lopez, C.A.; Uusitalo, J.J.; de Jong, D.H.; Gopal, S.M.; Periole, X.; Marrink, S.J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Pak, A.J.; Voth, G.A. Advances in coarse-grained modeling of macromolecular complexes. Curr. Opin. Struct. Biol. 2018, 52, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ekimoto, T.; Ikeguchi, M. Multiscale molecular dynamics simulations of rotary motor proteins. Biophys. Rev. 2018, 10, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Engkvist, O.; Norrby, P.O.; Selmi, N.; Lam, Y.H.; Peng, Z.; Sherer, E.C.; Amberg, W.; Erhard, T.; Smyth, L.A. Computational prediction of chemical reactions: Current status and outlook. Drug Discov. Today 2018, 23, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Miranda, W.E.; Ngo, V.A.; Perissinotti, L.L.; Noskov, S.Y. Computational membrane biophysics: From ion channel interactions with drugs to cellular function. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1643–1653. [Google Scholar] [CrossRef]
- Stevens, D.R.; Hammes-Schiffer, S. Exploring the Role of the Third Active Site Metal Ion in DNA Polymerase η with QM/MM Free Energy Simulations. J. Am. Chem. Soc. 2018, 140, 8965–8969. [Google Scholar] [CrossRef]
- Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 6 June 2019).
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Groban, E.S.; Narayanan, A.; Jacobson, M.P. Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput. Biol. 2006, 2, e32. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta 2015, 1850, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Parks, J.M.; Gumbart, J.C. Development of CHARMM-Compatible Force Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations. J. Chem. Theory Comput. 2018, 14, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, C.; Jing, Z.; Wu, C.; Piquemal, J.P.; Ponder, J.W.; Ren, P. AMOEBA Polarizable Atomic Multipole Force Field for Nucleic Acids. J. Chem. Theory Comput. 2018, 14, 2084–2108. [Google Scholar] [CrossRef]
- Zhang, C.; Bell, D.; Harger, M.; Ren, P. Polarizable Multipole-Based Force Field for Aromatic Molecules and Nucleobases. J. Chem. Theory Comput. 2017, 13, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P. FF12MC: A revised AMBER forcefield and new protein simulation protocol. Proteins 2016, 84, 1490–1516. [Google Scholar] [CrossRef]
- Xiong, X.; Chen, Z.; Cossins, B.P.; Xu, Z.; Shao, Q.; Ding, K.; Zhu, W.; Shi, J. Force fields and scoring functions for carbohydrate simulation. Carbohydr. Res. 2015, 12, 73–81. [Google Scholar] [CrossRef]
- Petrov, D.; Zagrovic, B. Are current atomistic force fields accurate enough to study proteins in crowded environments? PLoS Comput. Biol. 2014, 10, e1003638. [Google Scholar] [CrossRef]
- Riniker, S. Fixed-Charge Atomistic Force Fields for Molecular Dynamics Simulations in the Condensed Phase: An Overview. J. Chem. Inf. Model. 2018, 58, 565–578. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic validation of protein force fields against experimental data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef]
- Stone, J.E.; Hallock, M.J.; Phillips, J.C.; Peterson, J.R.; Luthey-Schulten, Z.; Schulten, K. Evaluation of Emerging Energy-Efficient Heterogeneous Computing Platforms for Biomolecular and Cellular Simulation Workloads. In Proceedings of the 30th IEEE International Parallel &Distributed Processing Symposium, Chicago, IL, USA, 24–27 May 2016; pp. 89–100. [Google Scholar]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Ikebe, J.; Umezawa, K.; Higo, J. Enhanced sampling simulations to construct free-energy landscape of protein-partner substrate interaction. Biophys. Rev. 2016, 8, 45–62. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Lin, X. Recent Advances in Computational Protocols Addressing Intrinsically Disordered Proteins. Biomolecules 2019, 9, 146. [Google Scholar] [CrossRef]
- Coskuner, O.; Wise-Scira, O. Structures and free energy landscapes of the A53T mutant-type α-synuclein protein and impact of A53T mutation on the structures of the wild-type α-synuclein protein with dynamics. ACS Chem. Neurosci. 2013, 4, 1101–1113. [Google Scholar] [CrossRef]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]
- Deserno, M. Mesoscopic membrane physics: Concepts, simulations, and selected applications. Macromol. Rapid Commun. 2009, 30, 752–771. [Google Scholar] [CrossRef]
- Davtyan, A.; Simunovic, M.; Voth, G.A. The mesoscopic membrane with proteins (MesM-P) model. J. Chem. Phys. 2017, 147, 044101. [Google Scholar] [CrossRef]
- Chavent, M.; Duncan, A.L.; Sansom, M.S. Molecular dynamics simulations of membrane proteins and their interactions: From nanoscale to mesoscale. Curr. Opin. Struct. Biol. 2016, 40, 8–16. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Clementi, C.; Nymeyer, H.; Onuchic, J.N. Topological and energetic factors: What determines the structural details of the transition state ensemble and ‘‘en-route’’ intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000, 298, 937–953. [Google Scholar] [CrossRef]
- Hyeon, C.; Dima, R.I.; Thirumalai, D. Pathways and kinetic barriers in mechanical unfolding and refolding of RNA and proteins. Structure 2006, 14, 1633–1645. [Google Scholar] [CrossRef]
- Hyeon, C.; Lorimer, G.H.; Thirumalai, D. Dynamics of allosteric transitions in GroEL. Proc. Natl. Acad. Sci. USA 2006, 103, 18939–18944. [Google Scholar] [CrossRef]
- Kolinski, A.; Skolnick, J. Reduced models of proteins and their applications. Polymer 2004, 45, 511–524. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)-round XII. Proteins Struct. Funct. Bioinform. 2018, 86, 7–15. [Google Scholar] [CrossRef]
- Liwo, A.; Baranowski, M.; Czaplewski, C.; Golas, E.; He, Y.; Jagiela, D.; Krupa, P.; Maciejczyk, M.; Makowski, M.; Mozolewska, M.A.; et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model. 2014, 20, 2306. [Google Scholar] [CrossRef]
- Yin, Y.; Sieradzan, A.K.; Liwo, A.; He, Y.; Scheraga, H.A. Physics-based potentials for coarse-grained modeling of protein-DNA interactions. J. Chem. Theory Comput. 2015, 11, 1792–1808. [Google Scholar] [CrossRef]
- Lu, Y.; Salsbury, F.R. Recapturing the Correlated Motions of Protein Using Coarse-Grained Models. Protein Pept. Lett. 2015, 22, 654–659. [Google Scholar] [CrossRef][Green Version]
- Delort, B.; Renault, P.; Charlier, L.; Raussin, F.; Martinez, J.; Floquet, N. Coarse-Grained Prediction of Peptide Binding to G-Protein Coupled Receptors. J. Chem. Inf. Model. 2017, 57, 562–571. [Google Scholar] [CrossRef]
- Hirano, R.; Yabuchi, T.; Sakurai, M.; Furuta, T. Development of an ATP force field for coarse grained simulation of ATPases and its application to the maltose transporter. J. Comput. Chem. 2019. [Google Scholar] [CrossRef]
- Damre, M.; Marchetto, A.; Giorgetti, A. MERMAID: Dedicated web server to prepare and run coarse-grained membrane protein dynamics. Nucleic Acids Res. 2019, 47, W456–W461. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, A.K.; Jakubowski, R. Introduction of steered molecular dynamics into UNRES coarse-grained simulations package. J. Comput. Chem. 2017, 38, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, E.; Krupa, P.; Mozolewska, M.A.; Liwo, A.; Kloczkowski, A. Reoptimized UNRES Potential for Protein Model Quality Assessment. Genes 2018, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, C.; Karczynska, A.; Sieradzan, A.K.; Liwo, A. UNRES server for physics-based coarse-grained simulations and prediction of protein structure, dynamics and thermodynamics. Nucleic Acids Res. 2018, 46, W304–W309. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Bogunia, M.; Mech, P.; Ganzynkowicz, R.; Giełdoń, A.; Liwo, A.; Makowski, M. Introduction of Phosphorylated Residues into the UNRES Coarse-Grained Model: Towards Modeling of Signaling Processes. J. Phys. Chem. B 2019, 123, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef]
- Kurcinski, M.; Oleniecki, T.; Ciemny, M.P.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2019, 35, 694–695. [Google Scholar] [CrossRef]
- Kar, P.; Gopal, S.M.; Cheng, Y.M.; Panahi, A.; Feig, M. Transferring the PRIMO Coarse-Grained Force Field to the Membrane Environment: Simulations of Membrane Proteins and Helix-Helix Association. J. Chem. Theory Comput. 2014, 10, 3459–3472. [Google Scholar] [CrossRef]
- Bouzakraoui, S.; Mousseau, N. Structural and thermodynamical properties of early human amylin oligomers using replica exchange molecular dynamics: Mutation effect of three key residues F15, H18 and F23. Phys. Chem. Chem. Phys. 2017, 19, 31290–31299. [Google Scholar] [CrossRef]
- Kynast, P.; Derreumaux, P.; Strodel, B. Evaluation of the coarse-grained OPEP force field for protein-protein docking. BMC Biophys. 2016, 9, 4. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; Sterpone, F.; Derreumaux, P. OPEP6: A New Constant-pH Molecular Dynamics Simulation Scheme with OPEP Coarse-Grained Force Field. J. Chem. Theory Comput. 2019, 15, 3875–3888. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Latshaw, D.C.; Hall, C.K. Aggregation of Aβ(17-36) in the Presence of Naturally Occurring Phenolic Inhibitors Using Coarse-Grained Simulations. J. Mol. Biol. 2017, 429, 3893–3908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hall, C.K. Seeding and cross-seeding fibrillation of N-terminal prion protein peptides PrP(120-144). Protein Sci. 2018, 27, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Calero-Rubio, C.; Paik, B.; Jia, X.; Kiick, K.L.; Roberts, C.J. Predicting unfolding thermodynamics and stable intermediates for alanine-rich helical peptides with the aid of coarse-grained molecular simulation. Biophys. Chem. 2016, 217, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Bereau, T.; Wang, Z.J.; Deserno, M. More than the sum of its parts: Coarse-grained peptide-lipid interactions from a simple cross-parametrization. J. Chem. Phys. 2014, 140, 115101. [Google Scholar] [CrossRef] [PubMed]
- Różycki, B.; Cazade, P.A.; O’Mahony, S.; Thompson, D.; Cieplak, M. The length but not the sequence of peptide linker modules exerts the primary influence on the conformations of protein domains in cellulosome multi-enzyme complexes. Phys. Chem. Chem. Phys. 2017, 19, 21414–21425. [Google Scholar] [CrossRef] [PubMed]
- Różycki, B.; Cieplak, M.; Czjzek, M. Large conformational fluctuations of the multi-domain xylanase Z of Clostridium thermocellum. J. Struct. Biol. 2015, 191, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Chalupska, D.; Różycki, B.; Humpolickova, J.; Faltova, L.; Klima, M.; Boura, E. Phosphatidylinositol 4-kinase IIIβ (PI4KB) forms highly flexible heterocomplexes that include ACBD3, 14-3-3, and Rab11 proteins. Sci. Rep. 2019, 9, 567. [Google Scholar] [CrossRef]
- Chalupska, D.; Eisenreichova, A.; Różycki, B.; Rezabkova, L.; Humpolickova, J.; Klima, M.; Boura, E. Structural analysis of phosphatidylinositol 4-kinase IIIβ (PI4KB)—14-3-3 protein complex reveals internal flexibility and explains 14-3-3 mediated protection from degradation in vitro. J. Struct. Biol. 2017, 200, 36–44. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef]
- Molinero, A.; Goddard, W.A. Goddard. M3B: A Coarse Grain Force Field for Molecular Simulations of Malto-Oligosaccharides and Their Water Mixtures. J. Phys. Chem. B 2004, 108, 1414–1427. [Google Scholar]
- Liu, P.; Izvekov, S.; Voth, G.A. Multiscale coarse-graining of monosaccharides. J. Phys. Chem. B 2007, 111, 11566–11575. [Google Scholar] [CrossRef] [PubMed]
- López, C.A.; Bellesia, G.; Redondo, A.; Langan, P.; Chundawat, S.P.; Dale, B.E.; Marrink, S.J.; Gnanakaran, S. MARTINI coarse-grained model for crystalline cellulose microfibers. J. Phys. Chem. B 2015, 119, 465–473. [Google Scholar] [CrossRef]
- Benner, S.W.; Hall, C.K. Development of a Coarse-Grained Model of Chitosan for Predicting Solution Behavior. J. Phys. Chem. B 2016, 120, 7253–7264. [Google Scholar] [CrossRef] [PubMed]
- Tsereteli, L.; Grafmüller, A. An accurate coarse-grained model for chitosan polysaccharides in aqueous solution. PLoS ONE 2017, 12, e0180938. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Cheng, X.; Smith, J.C. A Solvent-Free Coarse Grain Model for Crystalline and Amorphous Cellulose Fibrils. J. Chem. Theory Comput. 2011, 7, 2539–2548. [Google Scholar] [CrossRef]
- Srinivas, G.; Cheng, X.; Smith, J.C. Coarse-grain model for natural cellulose fibrils in explicit water. J. Phys. Chem. B 2014, 118, 3026–3034. [Google Scholar] [CrossRef]
- Glass, D.C.; Moritsugu, K.; Cheng, X.; Smith, J.C. REACH coarse-grained simulation of a cellulose fiber. Biomacromolecules 2012, 13, 2634–2644. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef]
- Negami, T.; Shimizu, K.; Terada, T. Coarse-grained molecular dynamics simulations of protein-ligand binding. J. Comput. Chem. 2014, 35, 1835–1845. [Google Scholar] [CrossRef]
- Thota, N.; Jiang, J. Self-assembly of amphiphilic peptide (AF)6H5K15 derivatives: Roles of hydrophilic and hydrophobic residues. J. Phys. Chem. B 2014, 118, 2683–2692. [Google Scholar] [CrossRef]
- Periole, X.; Knepp, A.M.; Sakmar, T.P.; Marrink, S.J.; Huber, T. Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J. Am. Chem. Soc. 2012, 134, 10959–10965. [Google Scholar] [CrossRef]
- Provasi, D.; Boz, M.B.; Johnston, J.M.; Filizola, M. Preferred supramolecular organization and dimer interfaces of opioid receptors from simulated self-association. PLoS Comput. Biol. 2015, 11, e1004148. [Google Scholar] [CrossRef]
- Sharma, S.; Juffer, A.H. An atomistic model for assembly of transmembrane domain of T cell receptor complex. J. Am. Chem. Soc. 2013, 135, 2188–2197. [Google Scholar] [CrossRef]
- Thallmair, S.; Ingólfsson, H.I.; Marrink, S.J. Cholesterol Flip-Flop Impacts Domain Registration in Plasma Membrane Models. J. Phys. Chem. Lett. 2018, 9, 5527–5533. [Google Scholar] [CrossRef]
- Li, S.; Wu, B.; Han, W. Parametrization of MARTINI for Modeling Hinging Motions in Membrane Proteins. J. Phys. Chem. B 2019, 123, 2254–2269. [Google Scholar] [CrossRef]
- UNRES web server. Available online: https://unres.pl/ (accessed on 25 May 2019).
- Sieradzan, A.K.; Lipska, A.G.; Lubecka, E.A. Shielding effect in protein folding. J. Mol. Graph. Model. 2018, 79, 118–132. [Google Scholar] [CrossRef]
- Yaşar, F.; Sieradzan, A.K.; Hansmann, U.H. Folding and self-assembly of a small heterotetramer. J. Chem. Phys. 2014, 140, 105103. [Google Scholar] [CrossRef]
- Krupa, P.; Mozolewska, M.A.; Joo, K.; Lee, J.; Czaplewski, C.; Liwo, A. Prediction of Protein Structure by Template-Based Modeling Combined with the UNRES Force Field. J. Chem. Inf. Model. 2015, 55, 1271–1281. [Google Scholar] [CrossRef]
- Karczyńska, A.S.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Liwo, A.; Czaplewski, C. Prediction of protein structure with the coarse-grained UNRES force field assisted by small X-ray scattering data and knowledge-based information. Proteins 2018, 86 (Suppl. 1), 228–239. [Google Scholar]
- Rojas, A.; Maisuradze, N.; Kachlishvili, K.; Scheraga, H.A.; Maisuradze, G.G. Elucidating Important Sites and the Mechanism for Amyloid Fibril Formation by Coarse-Grained Molecular Dynamics. ACS Chem. Neurosci. 2017, 8, 201–209. [Google Scholar] [CrossRef]
- Karczyńska, A.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Bojarski, K.K.; Zaborowski, B.; Liwo, A.; Ślusarz, R.; Ślusarz, M.; Lee, J.; et al. Use of the UNRES force field in template-assisted prediction of protein structures and the refinement of server models: Test with CASP12 targets. J. Mol. Graph. Model. 2018, 83, 92–99. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Giełdoń, A.; Yin, Y.; He, Y.; Scheraga, H.A.; Liwo, A. A new protein nucleic-acid coarse-grained force field based on the UNRES and NARES-2P force fields. J. Comput. Chem. 2018, 39, 2360–2370. [Google Scholar] [CrossRef]
- Lipska, A.G.; Seidman, S.R.; Sieradzan, A.K.; Giełdoń, A.; Liwo, A.; Scheraga, H.A. Molecular dynamics of protein A and a WW domain with a united-residue model including hydrodynamic interaction. J. Chem. Phys. 2016, 144, 184110. [Google Scholar] [CrossRef]
- Cossio, P.; Marinelli, F.; Laio, A.; Pietrucci, F. Optimizing the performance of bias-exchange metadynamics: Folding a 48-residue LysM domain using a coarse-grained model. J. Phys. Chem. B 2010, 114, 3259–3265. [Google Scholar] [CrossRef]
- Liwo, A.; Sieradzan, A.K.; Lipska, A.G.; Czaplewski, C.; Joung, I.; Żmudzińska, W.; Hałabis, A.; Ołdziej, S. A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. III. Determination of scale-consistent backbone-local and correlation potentials in the UNRES force field and force-field calibration and validation. J. Chem. Phys. 2019, 150, 155104. [Google Scholar]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar]
- Kmiecik, S.; Kolinski, A. Characterization of protein-folding pathways by reduced-space modeling. Proc. Natl. Acad. Sci. USA 2007, 104, 12330–12335. [Google Scholar] [CrossRef]
- Kmiecik, S.; Kolinski, A. Folding pathway of the b1 domain of protein G explored by multiscale modeling. Biophys. J. 2008, 94, 726–736. [Google Scholar] [CrossRef]
- Ciemny, M.P.; Badaczewska-Dawid, A.E.; Pikuzinska, M.; Kolinski, A.; Kmiecik, S. Modeling of Disordered Protein Structures Using Monte Carlo Simulations and Knowledge-Based Statistical Force Fields. Int. J. Mol. Sci. 2019, 20, 606. [Google Scholar] [CrossRef]
- Kmiecik, S.; Kolinski, A. Simulation of chaperonin effect on protein folding: A shift from nucleation-condensation to framework mechanism. J. Am. Chem. Soc. 2011, 133, 10283–10289. [Google Scholar] [CrossRef]
- Blaszczyk, M.; Jamroz, M.; Kmiecik, S.; Kolinski, A. CABS-fold: Server for the de novo and consensus-based prediction of protein structure. Nucleic Acids Res. 2013, 41, W406–W411. [Google Scholar] [CrossRef]
- Jamroz, M.; Kolinski, A.; Kmiecik, S. CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013, 41, W427–W431. [Google Scholar] [CrossRef]
- CABSflex. Available online: http://biocomp.chem.uw.edu.pl/CABSflex2 (accessed on 2 June 2019).
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef]
- Ciemny, M.P.; Debinski, A.; Paczkowska, M.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. Protein-peptide molecular docking with large-scale conformational changes: The p53-MDM2 interaction. Sci. Rep. 2016, 1, 37532. [Google Scholar] [CrossRef]
- Gopal, S.M.; Mukherjee, S.; Cheng, Y.M.; Feig, M. PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins 2010, 78, 1266–1281. [Google Scholar] [CrossRef]
- Kar, P.; Gopal, S.M.; Cheng, Y.M.; Predeus, A.; Feig, M. PRIMO: A Transferable Coarse-grained Force Field for Proteins. J. Chem. Theory Comput. 2013, 9, 3769–3788. [Google Scholar] [CrossRef]
- Derreumaux, P. From polypeptide sequences to structures using Monte Carlo simulations and an optimized potential. J. Chem. Phys. 1999, 111. [Google Scholar] [CrossRef]
- Sterpone, F.; Doutreligne, S.; Tran, T.T.; Melchionna, S.; Baaden, M.; Nguyen, P.H.; Derreumaux, P. Multi-scale simulations of biological systems using the OPEP coarse-grained model. Biochem. Biophys. Res. Commun. 2018, 498, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Chiricotto, M.; Melchionna, S.; Derreumaux, P.; Sterpone, F. Hydrodynamic effects on β-amyloid (16-22) peptide aggregation. J. Chem. Phys. 2016, 145, 035102. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.; Chang, I.; Hall, C.K. Extending the PRIME model for protein aggregation to all 20 amino acids. Proteins 2010, 78, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.; Chang, I.; Hall, C.K. Influence of temperature on formation of perfect tau fragment fibrils using PRIME20/DMD. Simul. Protein Sci. 2012, 21, 1514–1527. [Google Scholar] [CrossRef]
- Cheon, M.; Chang, I.; Hall, C.K. Spontaneous Formation of Twisted Aβ16-22 Fibrils in Large-Scale Molecular-Dynamics Simulations. Biophys. J. 2011, 101, 2493–2501. [Google Scholar] [CrossRef]
- Cheon, M.; Hall, C.K.; Chang, I. Structural Conversion of Aβ17-42 Peptides from Disordered Oligomers to U-Shape Protofilaments via Multiple Kinetic Pathways. PLoS Comput. Biol. 2015, 11, e1004258. [Google Scholar] [CrossRef]
- Latshaw, D.C.; Cheon, M.; Hall, C.K. Effects of macromolecular crowding on amyloid beta (16-22) aggregation using coarse-grained simulations. J. Phys. Chem. B 2014, 118, 13513–13526. [Google Scholar] [CrossRef]
- Latshaw, D.C., 2nd; Hall, C.K. Effects of hydrophobic macromolecular crowders onamyloid β(16-22) aggregation. Biophys. J. 2015, 109, 124–134. [Google Scholar] [CrossRef]
- Bunce, S.J.; Wang, Y.; Stewart, K.L.; Ashcroft, A.E.; Radford, S.E.; Hall, C.K.; Wilson, A.J. Molecular insights into the surface-catalyzed secondary nucleation of amyloid-β(40) (Aβ(40)) by the peptide fragment Aβ(16-22). Sci. Adv. 2019, 5. [Google Scholar] [CrossRef]
- Wang, Y.; Shao, Q.; Hall, C.K. N-terminal Prion Protein Peptides (PrP(120-144)) Form Parallel In-register β-Sheets via Multiple Nucleation-dependent Pathways. J. Biol. Chem. 2016, 291, 22093–22105. [Google Scholar] [CrossRef]
- Bereau, T.; Deserno, M. Generic coarse-grained model for protein folding and aggregation. J. Chem. Phys. 2009, 130, 235106. [Google Scholar] [CrossRef]
- Takada, S.; Luthey-Schulten, Z.; Wolynes, P.G. Folding dynamics with nonadditive forces: A simulation study of a designed helical protein and a random heteropolymer. J. Chem. Phys. 1999, 110, 11616–11629. [Google Scholar] [CrossRef][Green Version]
- Ding, F.; Borreguero, J.M.; Buldyrey, S.V.; Stanley, H.E.; Dokholyan, N.V. Mechanism for the alpha-helix to beta-hairpin transition. Proteins 2003, 53, 220–228. [Google Scholar] [CrossRef]
- Irbäck, A.; Sjunnesson, F.; Wallin, S. Three-helix-bundle protein in a Ramachandran model. Proc. Natl. Acad. Sci. USA 2000, 97, 13614–13618. [Google Scholar] [CrossRef]
- Miyazawa, S.; Jernigan, R.L. Estimation of Effective Interresidue Contact Energies from Protein Crystal-Structures—Quasi-Chemical Approximation. Macromolecules 1985, 18, 534–552. [Google Scholar] [CrossRef]
- Limbach, H.J.A.A.; Mann, B.A.; Holm, C. ESPRESSO-an extensible simulation package for research on soft matter systems. Comput. Phys. Commun. 2006, 174, 704–727. [Google Scholar] [CrossRef]
- Blanco, M.A.; Sahin, E.; Robinson, A.S.; Roberts, C.J. Coarse-grained model for colloidal protein interactions, B(22), and protein cluster formation. J. Phys. Chem. B 2013, 117, 16013–16028. [Google Scholar] [CrossRef]
- Kim, Y.C.; Hummer, G. Coarse-grained models for simulations of multiprotein complexes: Application to ubiquitin binding. J. Mol. Biol. 2008, 375, 1416–1433. [Google Scholar] [CrossRef]
- Różycki, B.; Kim, Y.C.; Hummer, G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure 2011, 19, 109–116. [Google Scholar] [CrossRef]
- Boura, E.; Rózycki, B.; Herrick, D.Z.; Chung, H.S.; Vecer, J.; Eaton, W.A.; Cafiso, D.S.; Hummer, G.; Hurley, J.H. Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 9437–9442. [Google Scholar] [CrossRef]
- Boura, E.; Różycki, B.; Chung, H.S.; Herrick, D.Z.; Canagarajah, B.; Cafiso, D.S.; Eaton, W.A.; Hummer, G.; Hurley, J.H. Solution structure of the ESCRT-I and -II supercomplex: Implications for membrane budding and scission. Structure 2012, 20, 874–886. [Google Scholar] [CrossRef]
- Leonard, T.A.; Różycki, B.; Saidi, L.F.; Hummer, G.; Hurley, J.H. Crystal structure and allosteric activation of protein kinase C βII. Cell 2011, 144, 55–66. [Google Scholar] [CrossRef]
- Francis, D.M.; Różycki, B.; Koveal, D.; Hummer, G.; Page, R.; Peti, W. Structural basis of p38α regulation by hematopoietic tyrosine phosphatase. Nat. Chem. Biol. 2011, 7, 916–924. [Google Scholar] [CrossRef]
- Francis, D.M.; Różycki, B.; Tortajada, A.; Hummer, G.; Peti, W.; Page, R. Resting and active states of the ERK2: HePTP complex. J. Am. Chem. Soc. 2011, 133, 17138–17141. [Google Scholar] [CrossRef]
- Steinkühler, J.; Różycki, B.; Alvey, C.; Lipowsky, R.; Weikl, T.R.; Dimova, R.; Discher, D.E. Membrane fluctuations and acidosis regulate cooperative binding of ‘marker of self’ protein CD47 with the macrophage checkpoint receptor SIRPα. J. Cell. Sci. 2018, 16. [Google Scholar] [CrossRef]
- Best, R.B.; Hummer, G. Coordinate-dependent diffusion in protein folding. Proc. Natl. Acad. Sci. USA 2010, 107, 1088–1093. [Google Scholar] [CrossRef]
- Kenzaki, H.; Koga, N.; Hori, N.; Kanada, R.; Li, W.; Okazaki, K.; Yao, X.Q.; Takada, S. CafeMol: A Coarse-Grained Biomolecular Simulator for Simulating Proteins at Work. J. Chem. Theory Comput. 2011, 7, 1979–1989. [Google Scholar] [CrossRef]
- Duus, J.; Gotfredsen, C.H.; Bock, K. Carbohydrate structural determination by NMR spectroscopy: Modern methods and limitations. Chem. Rev. 2000, 100, 4589–4614. [Google Scholar] [CrossRef]
- Yu, F.; Prestegard, J.H. Structural monitoring of oligosaccharides through 13C enrichment and NMR observation of acetyl groups. Biophys. J. 2006, 91, 1952–1959. [Google Scholar] [CrossRef][Green Version]
- Wormald, M.R.; Petrescu, A.J.; Pao, Y.L.; Glithero, A.; Elliott, T.; Dwek, R.A. Conformational studies of oligosaccharides and glycopeptides: Complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 2002, 102, 371–386. [Google Scholar] [CrossRef]
- Bellesia, G.; Chundawat, S.P.; Langan, P.; Redondo, A.; Dale, B.E.; Gnanakaran, S. Gnanakaran. Coarse-Grained Model for the Interconversion between Native and Liquid Ammonia-Treated Crystalline Cellulose. J. Phys. Chem. B 2012, 116, 8031–8037. [Google Scholar] [CrossRef]
- López, C.A.; Rzepiela, A.J.; de Vries, A.H.; Dijkhuizen, L.; Hünenberger, P.H.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to Carbohydrates. J. Chem. Theory Comput. 2009, 5, 3195–3210. [Google Scholar] [CrossRef]
- Gu, R.X.; Ingólfsson, H.I.; de Vries, A.H.; Marrink, S.J.; Tieleman, D.P. Ganglioside—Lipid and Ganglioside-Protein Interactions Revealed by Coarse-Grained and Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3262–3275. [Google Scholar] [CrossRef]
- Yu, Z.; Lau, D. Development of a coarse-grained α-chitin model on the basis of MARTINI forcefield. J. Mol. Model. 2015, 21, 128. [Google Scholar] [CrossRef]
- Schmalhorst, P.S.; Deluweit, F.; Scherrers, R.; Heisenberg, C.P.; Sikora, M. Overcoming the Limitations of the MARTINI Force Field in Simulations of Polysaccharides. J. Chem. Theory Comput. 2017, 13, 5039–5053. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Polysaccharide-Protein Complexes in a Coarse-Grained Model. J. Phys. Chem. B 2015, 119, 12028–12041. [Google Scholar] [CrossRef]
- Grime, J.M.; Dama, J.F.; Ganser-Pornillos, B.K.; Woodward, C.L.; Jensen, G.J.; Yeager, M.; Voth, G.A. Coarse-grained simulation reveals key features of HIV-1 capsid self-assembly. Nat. Commun. 2016, 7, 11568. [Google Scholar] [CrossRef]
- Pak, A.J.; Grime, J.M.A.; Sengupta, P.; Chen, A.K.; Durumeric, A.E.P.; Srivastava, A.; Yeager, M.; Briggs, J.A.G.; Lippincott-Schwartz, J.; Voth, G.A. Immature HIV-1 lattice assembly dynamics are regulated by scaffolding from nucleic acid and the plasma membrane. Proc. Natl. Acad. Sci. USA 2017, 114, E10056–E10065. [Google Scholar] [CrossRef]
- Qiao, X.; Jeon, J.; Weber, J.; Zhu, F.; Chen, B. Mechanism of polymorphism and curvature of HIV capsid assemblies probed by 3D simulations with a novel coarse grain model. Biochim. Biophys. Acta 2015, 1850, 2353–2367. [Google Scholar] [CrossRef]
- Grime, J.M.; Voth, G.A. Early stages of the HIV-1 capsid protein lattice formation. Biophys. J. 2012, 103, 1774–1783. [Google Scholar] [CrossRef]
- Qiao, X.; Jeon, J.; Weber, J.; Zhu, F.; Chen, B. Construction of a novel coarse grain model for simulations of HIV capsid assembly to capture the backbone structure and inter-domain motions in solution. Data Brief. 2015, 5, 506–512. [Google Scholar] [CrossRef][Green Version]
- Charlier, L.; Louet, M.; Chaloin, L.; Fuchs, P.; Martinez, J.; Muriaux, D.; Favard, C.; Floquet, N. Coarse-grained simulations of the HIV-1 matrix protein anchoring: Revisiting its assembly on membrane domains. Biophys. J. 2014, 106, 577–585. [Google Scholar] [CrossRef]
- Chen, B.; Tycko, R. Simulated self-assembly of the HIV-1 capsid: Protein shape and native contacts are sufficient for two-dimensional lattice formation. Biophys. J. 2011, 100, 3035–3044. [Google Scholar] [CrossRef]
- Mugnai, M.L.; Thirumalai, D. Kinematics of the lever arm swing in myosin VI. Proc. Natl. Acad. Sci. USA 2017, 114, E4389–E4398. [Google Scholar] [CrossRef]
- Zhang, Z.; Thirumalai, D. Dissecting the kinematics of the kinesin step. Structure 2012, 20, 628–640. [Google Scholar] [CrossRef]
- Sułkowska, J.I.; Sułkowski, P.; Onuchic, J. Dodging the crisis of folding proteins with knots. Proc. Natl. Acad. Sci. USA 2009, 106, 3119–3124. [Google Scholar] [CrossRef]
- Terakawa, T.; Takada, S. Multiscale ensemble modeling of intrinsically disordered proteins: p53 N-terminal domain. Biophys. J. 2011, 101, 1450–1458. [Google Scholar] [CrossRef]
- Ramis, R.; Ortega-Castro, J.; Casasnovas, R.; Mariño, L.; Vilanova, B.; Adrover, M.; Frau, J. A Coarse-Grained Molecular Dynamics Approach to the Study of the Intrinsically Disordered Protein α-Synuclein. J. Chem. Inf. Model. 2019, 59, 1458–1471. [Google Scholar] [CrossRef]
- Poma, A.B.; Guzman, H.V.; Li, M.S.; Theodorakis, P.E. Mechanical and thermodynamic properties of Aβ(42), Aβ(40), and α-synuclein fibrils: A coarse-grained method to complement experimental studies. Beilstein J. Nanotechnol. 2019, 10, 500–513. [Google Scholar] [CrossRef]
- Ilie, I.M.; den Otter, W.K.; Briels, W.J. The attachment of α-synuclein to a fiber: A coarse-grain approach. J. Chem. Phys. 2017, 146, 115102. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Giorgetti, A.; Carloni, P. Multiscale simulations on human Frizzled and Taste2 GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 8–16. [Google Scholar] [CrossRef]
- Tarenzi, T.; Calandrini, V.; Potestio, R.; Carloni, P. Open-Boundary Molecular Mechanics/Coarse-Grained Framework for Simulations of Low-Resolution G-Protein-Coupled Receptor-Ligand Complexes. J. Chem. Theory Comput. 2019, 15, 2101–2109. [Google Scholar] [CrossRef]
- Shimizu, M.; Takada, S. Reconstruction of Atomistic Structures from Coarse-Grained Models for Protein-DNA Complexes. J. Chem. Theory Comput. 2018, 14, 1682–1694. [Google Scholar] [CrossRef]
- Lombardi, L.E.; Martí, M.A.; Capece, L. CG2AA: Backmapping protein coarse-grained structures. Bioinformatics 2016, 32, 1235–1237. [Google Scholar] [CrossRef]
- Sterpone, F.; Derreumaux, P.; Melchionna, S. Protein Simulations in Fluids: Coupling the OPEP Coarse-Grained Force Field with Hydrodynamics. J. Chem. Theory Comput. 2015, 11, 1843–1853. [Google Scholar] [CrossRef]
- Sterpone, F.; Derreumaux, P.; Melchionna, S. Molecular Mechanism of Protein Unfolding under Shear: A Lattice Boltzmann Molecular Dynamics Study. J. Phys. Chem. B 2018, 122, 1573–1579. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, C.; Ma, R.; Zhang, Z. Backmapping from Multiresolution Coarse-Grained Models to Atomic Structures of Large Biomolecules by Restrained Molecular Dynamics Simulations Using Bayesian Inference. J. Chem. Theory Comput. 2019, 15, 3344–3353. [Google Scholar] [CrossRef]
- Fritz, D.; Koschke, K.; Harmandarism, V.A.; van der Vegt, N.F.; Kremer, K. Multiscale modeling of soft matter: Scaling of dynamics. Phys. Chem. Chem. Phys. 2011, 13, 10412–10420. [Google Scholar] [CrossRef]
- Rudzinski, J.F.; Noid, W.G. Coarse-graining entropy, forces, and structures. J. Chem. Phys. 2011, 135, 214101. [Google Scholar] [CrossRef]
- Zhe, W.; Qiang, C.; Yethiraj, A. Driving Force for the Association of Hydrophobic Peptides: The Importance of Electrostatic Interactions in Coarse-Grained Water Models. J. Phys. Chem. Lett. 2011, 2, 1794–1798. [Google Scholar]
- Noid, W.G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139, 090901. [Google Scholar] [CrossRef]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Tamò, G.E.; Abriata, L.A.; Dal Peraro, M. The importance of dynamics in integrative modeling of supramolecular assemblies. Curr. Opin. Struct. Biol. 2015, 31, 28–34. [Google Scholar] [CrossRef]
- Feig, M.; Sugita, Y. Reaching new levels of realism in modeling biological macromolecules in cellular environments. J. Mol. Graph. Model. 2013, 45, 144–156. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Coarse-Grained Models | Granularity of the Model | Recent Advances, Example Application and Additional Information |
|---|---|---|
| Proteins | ||
| MARTINI | Up to five beads per amino acid residue | mechanism of allostery [40], peptide binding to GPCRs [41], Parameters developed for ATP molecule [42] MERMAID (webserver for simulation of membrane proteins (http://molsim.sci.univr.it/mangesh/index.php) [43] |
| UNRES | Two beads per residue | Steered molecular dynamics integrated to UNRES [44], deep feed-forward neural network-based re-optimization of UNRES for ranking of protein structure models [45], freely accessible server launched (http://unres-server.chem.ug.edu.pl.) [46], parameters developed for phosphorylated residues [47] |
| CABS | Four beads per residue | CABS-dock (webserver for flexible docking of peptides) [48] CABS-flex standalone package [49], |
| PRIMO | Three to eight beads per residue | Provides high resolution and transferability [50] |
| OPEP | Up to six beads per residue | Replica Exchange MD and OPEP [51], protein–protein docking [52], OPEP 6 (Constant-pH Molecular Dynamics Simulation Scheme) [53]; |
| PRIME | Four beads per residue | Effect of inhibitors on Aβ fibril formation [54]; co-aggregation of Aβ40 and Aβ16–22 peptides cross seeding in fibrillation of prion protein peptides [55] |
| Bereau and Deserno | Four beads per residue | Refined model for thermodynamics of unfolding process of peptides [56] cross parameterization of two models to study peptide-membrane [57] |
| Kim and Hummer | Single bead centered at cα atom. | Study of multi-domain cellulosomes [58,59]; coupled with SAXS to study highly flexible protein complexes [60,61]; phase behavior of intrinsically disordered proteins [62]. |
| Carbohydrates | ||
| M3B | Three beads per monosaccharide | Pioneer model for CG methods of carbohydrates [63] |
| Bellesia model | Five beads per monosaccharide | Developed to study the structural transition from cellulose Iβ to cellulose III(I) [64] |
| MARTINI | Three beads per monosaccharide | physicochemical properties of cellulose Iβ [65]; Chitosan and solution behavior [66]; self-assembly of polysaccharide [67] |
| Srinivas model | Single bead per monosaccharide | Solvent free coarse-grained model [68]; study of cellulose fibrils [69] |
| REACH | Single bead per monosaccharide | Developed to study the elastic properties of the cellulose fibril [70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Li, W. Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications. Int. J. Mol. Sci. 2019, 20, 3774. https://doi.org/10.3390/ijms20153774
Singh N, Li W. Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications. International Journal of Molecular Sciences. 2019; 20(15):3774. https://doi.org/10.3390/ijms20153774
Chicago/Turabian StyleSingh, Nidhi, and Wenjin Li. 2019. "Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications" International Journal of Molecular Sciences 20, no. 15: 3774. https://doi.org/10.3390/ijms20153774
APA StyleSingh, N., & Li, W. (2019). Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications. International Journal of Molecular Sciences, 20(15), 3774. https://doi.org/10.3390/ijms20153774