The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases


Abstract
1. Introduction
2. Important Role of HO-1 in Atherosclerosis
3. Mechanistic Actions of HO-1 in Oxidative Stress and Inflammation
4. HO-1 Expression in Atherosclerotic Diseases States (Animal Studies)
4.1. Myocardial Infarction
4.2. Heart Failure
5. HO-1 Expression in Patients with Atherosclerotic Diseases (Clinical Studies)
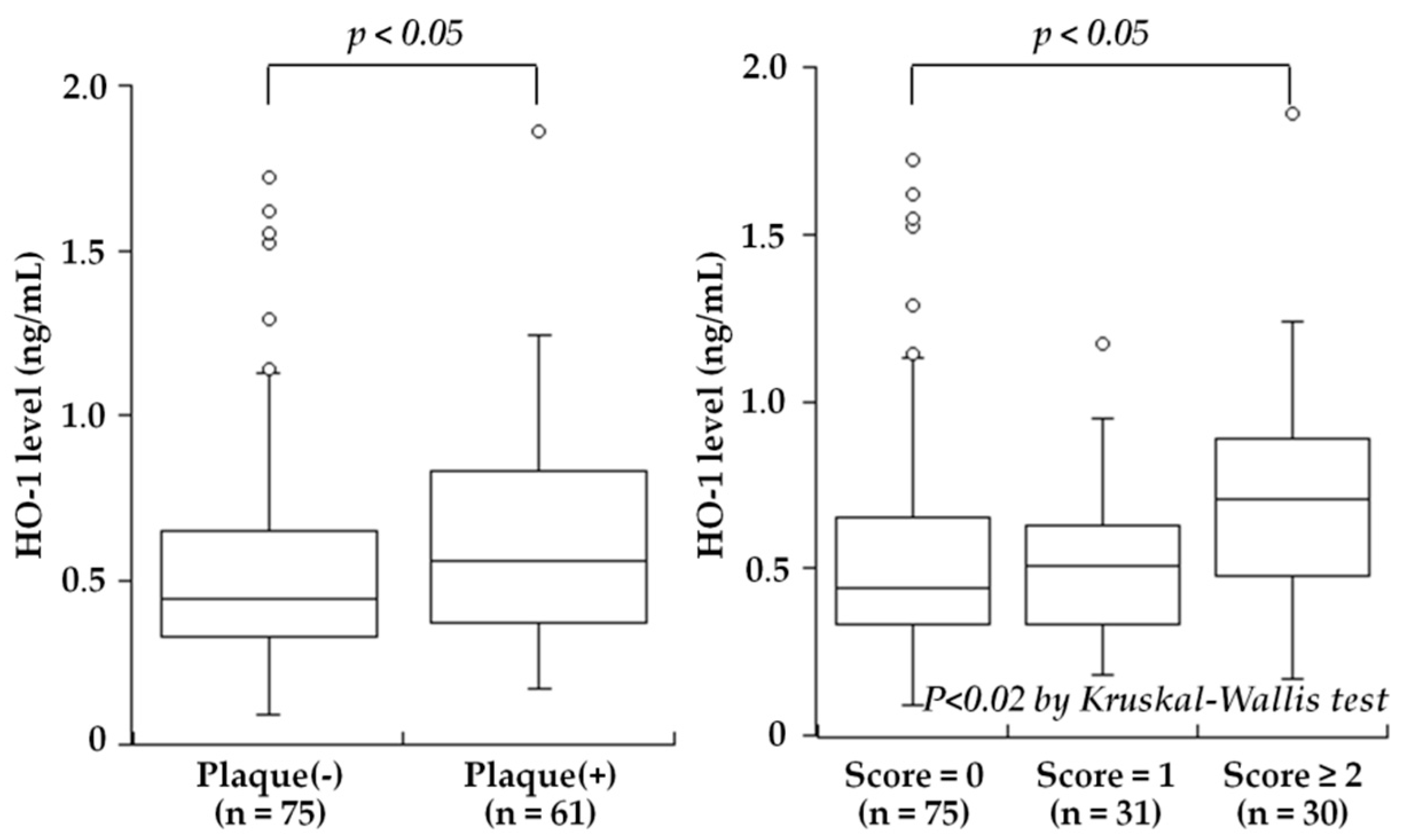
5.1. Carotid Atherosclerosis
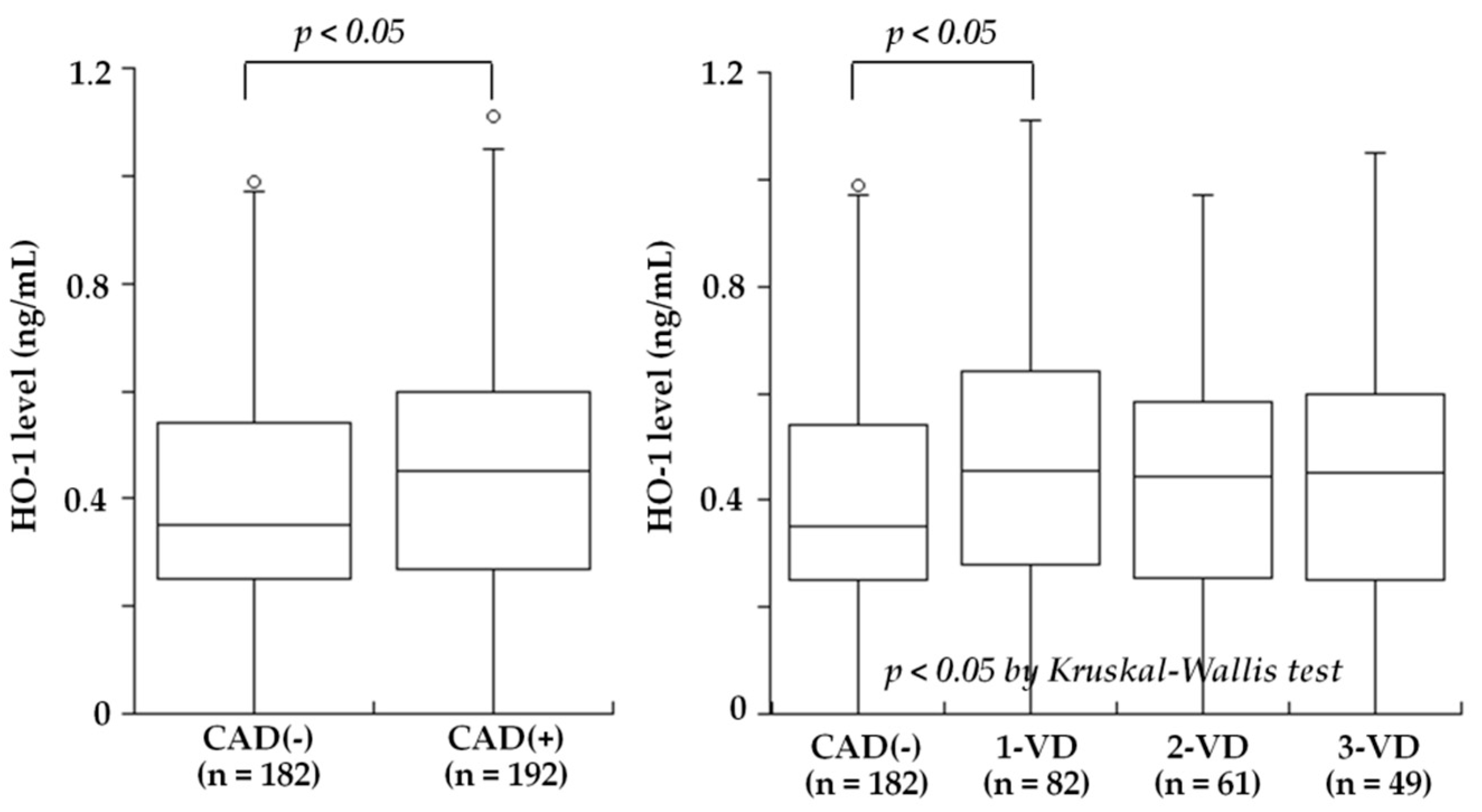
5.2. Coronary Artery Disease (CAD) and Peripheral Artery Disease (PAD)
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| HO | Heme oxygenase |
| CO | Carbon monoxide |
| CAD | Coronary artery disease |
| PAD | Peripheral artery disease |
| LDL | Low-density lipoprotein |
| UGT1A1 | UDP-glucuronosyltransferase 1A1 |
| PPAR-α | Peroxisome proliferator-activated receptor-alpha |
| MCP-1 | Monocyte chemoattractant protein-1 |
| IL-6 | Interleukin-6 |
| SR-A | Scavenger receptor-A |
| AMI | Acute myocardial infarction |
| ABI | Ankle-brachial index |
| CI | Confidence interval |
| SAP | Stable angina pectoris |
| UAP | Unstable angina pectoris |
| ACS | Acute coronary syndrome |
| TIA | Transient ischemic attack |
References
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [PubMed]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a target for therapeutic interventions. Front. Pharmacol. 2017, 8, 146. [Google Scholar] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [PubMed]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [PubMed]
- Fredenburgh, L.E.; Merz, A.A.; Cheng, S. Haeme oxygenase signalling pathway: Implications for cardiovascular disease. Eur. Heart J. 2015, 36, 1512–1518. [Google Scholar] [PubMed]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Ann. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar]
- Wu, M.L.; Ho, Y.C.; Lin, C.Y.; Yet, S.F. Heme oxygenase-1 in inflammation and cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 150–158. [Google Scholar]
- Yoshida, T.; Biro, P.; Cohen, T.; Muller, R.M.; Shibahara, S. Human heme oxygenase cdna and induction of its mrna by hemin. Eur. J. Biochem. 1988, 171, 457–461. [Google Scholar]
- Alam, J.; Shibahara, S.; Smith, A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J. Biol. Chem. 1989, 264, 6371–6375. [Google Scholar]
- Keyse, S.M.; Tyrrell, R.M. Induction of the heme oxygenase gene in human skin fibroblasts by hydrogen peroxide and uva (365 nm) radiation: Evidence for the involvement of the hydroxyl radical. Carcinogenesis 1990, 11, 787–791. [Google Scholar]
- Cantoni, L.; Rossi, C.; Rizzardini, M.; Gadina, M.; Ghezzi, P. Interleukin-1 and tumour necrosis factor induce hepatic haem oxygenase. Feedback regulation by glucocorticoids. Biochem. J. 1991, 279, 891–894. [Google Scholar] [PubMed]
- Rizzardini, M.; Carelli, M.; Cabello Porras, M.R.; Cantoni, L. Mechanisms of endotoxin-induced haem oxygenase mrna accumulation in mouse liver: Synergism by glutathione depletion and protection by n-acetylcysteine. Biochem. J. 1994, 304, 477–483. [Google Scholar] [PubMed]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [PubMed]
- Wang, L.J.; Lee, T.S.; Lee, F.Y.; Pai, R.C.; Chau, L.Y. Expression of heme oxygenase-1 in atherosclerotic lesions. Am. J. Pathol. 1998, 152, 711–720. [Google Scholar] [PubMed]
- Chen, S.M.; Li, Y.G.; Wang, D.M. Study on changes of heme oxygenase-1 expression in patients with coronary heart disease. Clin. Cardiol. 2005, 28, 197–201. [Google Scholar] [PubMed]
- Ameriso, S.F.; Villamil, A.R.; Zedda, C.; Parodi, J.C.; Garrido, S.; Sarchi, M.I.; Schultz, M.; Boczkowski, J.; Sevlever, G.E. Heme oxygenase-1 is expressed in carotid atherosclerotic plaques infected by helicobacter pylori and is more prevalent in asymptomatic subjects. Stroke 2005, 36, 1896–1900. [Google Scholar]
- Ijas, P.; Nuotio, K.; Saksi, J.; Soinne, L.; Saimanen, E.; Karjalainen-Lindsberg, M.L.; Salonen, O.; Sarna, S.; Tuimala, J.; Kovanen, P.T.; et al. Microarray analysis reveals overexpression of cd163 and ho-1 in symptomatic carotid plaques. Arterioscler. Thromb Vasc. Biol 2007, 27, 154–160. [Google Scholar]
- Brydun, A.; Watari, Y.; Yamamoto, Y.; Okuhara, K.; Teragawa, H.; Kono, F.; Chayama, K.; Oshima, T.; Ozono, R. Reduced expression of heme oxygenase-1 in patients with coronary atherosclerosis. Hypertens. Res. 2007, 30, 341–348. [Google Scholar]
- Cheng, C.; Noordeloos, A.M.; Jeney, V.; Soares, M.P.; Moll, F.; Pasterkamp, G.; Serruys, P.W.; Duckers, H.J. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 2009, 119, 3017–3027. [Google Scholar]
- Idriss, N.K.; Lip, G.Y.; Balakrishnan, B.; Jaumdally, R.; Boos, C.J.; Blann, A.D. Plasma haemoxygenase-1 in coronary artery disease. A comparison with angiogenin, matrix metalloproteinase-9, tissue inhibitor of metalloproteinase-1 and vascular endothelial growth factor. Thromb. Haemost. 2010, 104, 1029–1037. [Google Scholar]
- Novo, G.; Cappello, F.; Rizzo, M.; Fazio, G.; Zambuto, S.; Tortorici, E.; Marino Gammazza, A.; Corrao, S.; Zummo, G.; De Macario, E.C.; et al. Hsp60 and heme oxygenase-1 (hsp32) in acute myocardial infarction. Transl. Res. 2011, 157, 285–292. [Google Scholar] [PubMed]
- Yunoki, K.; Inoue, T.; Sugioka, K.; Nakagawa, M.; Inaba, M.; Wada, S.; Ohsawa, M.; Komatsu, R.; Itoh, A.; Haze, K.; et al. Association between hemoglobin scavenger receptor and heme oxygenase-1-related anti-inflammatory mediators in human coronary stable and unstable plaques. Hum. Pathol. 2013, 44, 2256–2265. [Google Scholar] [PubMed]
- Li, X.; Song, G.; Jin, Y.; Liu, H.; Li, C.; Han, C.; Ren, S. Higher level of heme oxygenase-1 in patients with stroke than tia. J. Thorac. Dis. 2014, 6, 772–777. [Google Scholar] [PubMed]
- Signorelli, S.S.; Li Volsi, G.; Fiore, V.; Mangiafico, M.; Barbagallo, I.; Parenti, R.; Rizzo, M.; Li Volti, G. Plasma heme oxygenase-1 is decreased in peripheral artery disease patients. Mol. Med. Rep. 2016, 14, 3459–3463. [Google Scholar] [PubMed]
- Kishimoto, Y.; Sasaki, K.; Saita, E.; Niki, H.; Ohmori, R.; Kondo, K.; Momiyama, Y. Plasma heme oxygenase-1 levels and carotid atherosclerosis. Stroke 2018, 49, 2230–2232. [Google Scholar] [PubMed]
- Kishimoto, Y.; Ibe, S.; Saita, E.; Sasaki, K.; Niki, H.; Miura, K.; Ikegami, Y.; Ohmori, R.; Kondo, K.; Momiyama, Y. Plasma heme oxygenase-1 levels in patients with coronary and peripheral artery diseases. Dis. Markers 2018, 2018, 6138124. [Google Scholar] [PubMed]
- Fiorelli, S.; Porro, B.; Cosentino, N.; Di Minno, A.; Manega, C.M.; Fabbiocchi, F.; Niccoli, G.; Fracassi, F.; Barbieri, S.; Marenzi, G.; et al. Activation of nrf2/ho-1 pathway and human atherosclerotic plaque vulnerability:An in vitro and in vivo study. Cells 2019, 8, 356. [Google Scholar]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [PubMed]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [PubMed]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [PubMed]
- Ishikawa, K.; Navab, M.; Lusis, A.J. Vasculitis, atherosclerosis, and altered hdl composition in heme-oxygenase-1-knockout mice. Int. J. Hypertens. 2012, 2012, 948203. [Google Scholar] [PubMed]
- Agarwal, A.; Balla, J.; Balla, G.; Croatt, A.J.; Vercellotti, G.M.; Nath, K.A. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized ldl. Am. J. Physiol. 1996, 271, F814–F823. [Google Scholar] [PubMed]
- Yamaguchi, M.; Sato, H.; Bannai, S. Induction of stress proteins in mouse peritoneal macrophages by oxidized low-density lipoprotein. Biochem. Biophys. Res. Commun. 1993, 193, 1198–1201. [Google Scholar] [PubMed]
- Ishikawa, K.; Navab, M.; Leitinger, N.; Fogelman, A.M.; Lusis, A.J. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized ldl. J. Clin. Investig. 1997, 100, 1209–1216. [Google Scholar]
- Ishikawa, K.; Sugawara, D.; Wang, X.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme oxygenase-1 inhibits atherosclerotic lesion formation in ldl-receptor knockout mice. Circ. Res. 2001, 88, 506–512. [Google Scholar]
- Ishikawa, K.; Sugawara, D.; Goto, J.; Watanabe, Y.; Kawamura, K.; Shiomi, M.; Itabe, H.; Maruyama, Y. Heme oxygenase-1 inhibits atherogenesis in watanabe heritable hyperlipidemic rabbits. Circulation 2001, 104, 1831–1836. [Google Scholar]
- Juan, S.H.; Lee, T.S.; Tseng, K.W.; Liou, J.Y.; Shyue, S.K.; Wu, K.K.; Chau, L.Y. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein e-deficient mice. Circulation 2001, 104, 1519–1525. [Google Scholar]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar]
- Qiao, H.; Sai, X.; Gai, L.; Huang, G.; Chen, X.; Tu, X.; Ding, Z. Association between heme oxygenase 1 gene promoter polymorphisms and susceptibility to coronary artery disease: A huge review and meta-analysis. Am. J. Epidemiol. 2014, 179, 1039–1048. [Google Scholar]
- Yamada, N.; Yamaya, M.; Okinaga, S.; Nakayama, K.; Sekizawa, K.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am. J. Hum. Genet. 2000, 66, 187–195. [Google Scholar] [PubMed]
- Ono, K.; Goto, Y.; Takagi, S.; Baba, S.; Tago, N.; Nonogi, H.; Iwai, N. A promoter variant of the heme oxygenase-1 gene may reduce the incidence of ischemic heart disease in japanese. Atherosclerosis 2004, 173, 315–319. [Google Scholar] [PubMed]
- Schillinger, M.; Exner, M.; Mlekusch, W.; Domanovits, H.; Huber, K.; Mannhalter, C.; Wagner, O.; Minar, E. Heme oxygenase-1 gene promoter polymorphism is associated with abdominal aortic aneurysm. Thromb. Res. 2002, 106, 131–136. [Google Scholar] [PubMed]
- Kaneda, H.; Ohno, M.; Taguchi, J.; Togo, M.; Hashimoto, H.; Ogasawara, K.; Aizawa, T.; Ishizaka, N.; Nagai, R. Heme oxygenase-1 gene promoter polymorphism is associated with coronary artery disease in japanese patients with coronary risk factors. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1680–1685. [Google Scholar] [PubMed]
- Funk, M.; Endler, G.; Schillinger, M.; Mustafa, S.; Hsieh, K.; Exner, M.; Lalouschek, W.; Mannhalter, C.; Wagner, O. The effect of a promoter polymorphism in the heme oxygenase-1 gene on the risk of ischaemic cerebrovascular events: The influence of other vascular risk factors. Thromb. Res. 2004, 113, 217–223. [Google Scholar] [PubMed]
- Chen, Y.H.; Chau, L.Y.; Chen, J.W.; Lin, S.J. Serum bilirubin and ferritin levels link heme oxygenase-1 gene promoter polymorphism and susceptibility to coronary artery disease in diabetic patients. Diabetes Care 2008, 31, 1615–1620. [Google Scholar] [PubMed]
- Chen, M.; Zhou, L.; Ding, H.; Huang, S.; He, M.; Zhang, X.; Cheng, L.; Wang, D.; Hu, F.B.; Wu, T. Short (gt) ( n ) repeats in heme oxygenase-1 gene promoter are associated with lower risk of coronary heart disease in subjects with high levels of oxidative stress. Cell Stress Chaperones 2012, 17, 329–338. [Google Scholar] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An nrf2/small maf heterodimer mediates the induction of phase ii detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [PubMed]
- Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative stress in cardiovascular diseases: Involvement of nrf2 antioxidant redox signaling in macrophage foam cells formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. Nrf2 and nf-b interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [PubMed]
- Mayer, M. Association of serum bilirubin concentration with risk of coronary artery disease. Clin. Chem. 2000, 46, 1723–1727. [Google Scholar] [PubMed]
- Boon, A.C.; Hawkins, C.L.; Bisht, K.; Coombes, J.S.; Bakrania, B.; Wagner, K.H.; Bulmer, A.C. Reduced circulating oxidized ldl is associated with hypocholesterolemia and enhanced thiol status in gilbert syndrome. Free Radic. Biol. Med. 2012, 52, 2120–2127. [Google Scholar] [PubMed]
- Stojanov, M.; Stefanovic, A.; Dzingalasevic, G.; Ivanisevic, J.; Miljkovic, M.; Mandic-Radic, S.; Prostran, M. Total bilirubin in young men and women: Association with risk markers for cardiovascular diseases. Clin. Biochem. 2013, 46, 1516–1519. [Google Scholar] [PubMed]
- Nascimento, H.; Alves, A.I.; Coimbra, S.; Catarino, C.; Gomes, D.; Bronze-da-Rocha, E.; Costa, E.; Rocha-Pereira, P.; Aires, L.; Mota, J.; et al. Bilirubin is independently associated with oxidized ldl levels in young obese patients. Diabetol. Metab. Syndr. 2015, 7, 4. [Google Scholar] [PubMed]
- Schwertner, H.A.; Jackson, W.G.; Tolan, G. Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem. 1994, 40, 18–23. [Google Scholar] [PubMed]
- Turfan, M.; Duran, M.; Poyraz, F.; Yayla, C.; Akboga, M.K.; Sahinarslan, A.; Tavil, Y.; Pasaoglu, H.; Boyaci, B. Inverse relationship between serum total bilirubin levels and severity of disease in patients with stable coronary artery disease. Coron. Artery Dis. 2013, 24, 29–32. [Google Scholar]
- Kang, S.J.; Kim, D.; Park, H.E.; Chung, G.E.; Choi, S.H.; Choi, S.Y.; Lee, W.; Kim, J.S.; Cho, S.H. Elevated serum bilirubin levels are inversely associated with coronary artery atherosclerosis. Atherosclerosis 2013, 230, 242–248. [Google Scholar]
- Akboga, M.K.; Canpolat, U.; Sahinarslan, A.; Alsancak, Y.; Nurkoc, S.; Aras, D.; Aydogdu, S.; Abaci, A. Association of serum total bilirubin level with severity of coronary atherosclerosis is linked to systemic inflammation. Atherosclerosis 2015, 240, 110–114. [Google Scholar]
- Lin, J.P.; Vitek, L.; Schwertner, H.A. Serum bilirubin and genes controlling bilirubin concentrations as biomarkers for cardiovascular disease. Clin. Chem. 2010, 56, 1535–1543. [Google Scholar]
- Stender, S.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Grande, P.; Tybjaerg-Hansen, A. Genetically elevated bilirubin and risk of ischaemic heart disease: Three mendelian randomization studies and a meta-analysis. J. Intern. Med. 2013, 273, 59–68. [Google Scholar] [PubMed]
- Kobayashi, A.; Ishikawa, K.; Matsumoto, H.; Kimura, S.; Kamiyama, Y.; Maruyama, Y. Synergetic antioxidant and vasodilatory action of carbon monoxide in angiotensin ii - induced cardiac hypertrophy. Hypertension 2007, 50, 1040–1048. [Google Scholar] [PubMed]
- Taille, C.; El-Benna, J.; Lanone, S.; Dang, M.C.; Ogier-Denis, E.; Aubier, M.; Boczkowski, J. Induction of heme oxygenase-1 inhibits nad(p)h oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J. Biol. Chem. 2004, 279, 28681–28688. [Google Scholar] [PubMed]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational significance of heme oxygenase in obesity and metabolic syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [PubMed]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; Shapiro, J.; Abraham, N.G.; Kappas, A. Increased ho-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of fgf21. Obesity (Silver Spring) 2014, 22, 705–712. [Google Scholar]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin binding to pparalpha inhibits lipid accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar]
- Gordon, D.M.; Blomquist, T.M.; Miruzzi, S.A.; McCullumsmith, R.; Stec, D.E.; Hinds, T.D., Jr. Rna sequencing in human hepg2 hepatocytes reveals ppar-alpha mediates transcriptome responsiveness of bilirubin. Physiol. Genom. 2019, 51, 234–240. [Google Scholar]
- Kawamura, K.; Ishikawa, K.; Wada, Y.; Kimura, S.; Matsumoto, H.; Kohro, T.; Itabe, H.; Kodama, T.; Maruyama, Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 155–160. [Google Scholar] [PubMed]
- Kim, J.A.; Territo, M.C.; Wayner, E.; Carlos, T.M.; Parhami, F.; Smith, C.W.; Haberland, M.E.; Fogelman, A.M.; Berliner, J.A. Partial characterization of leukocyte binding molecules on endothelial cells induced by minimally oxidized ldl. Arterioscler. Thromb. 1994, 14, 427–433. [Google Scholar]
- Zakkar, M.; Van der Heiden, K.; Luong le, A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar]
- Kim, M.; Kim, S.; Lim, J.H.; Lee, C.; Choi, H.C.; Woo, C.H. Laminar flow activation of erk5 protein in vascular endothelium leads to atheroprotective effect via nf-e2-related factor 2 (nrf2) activation. J. Biol. Chem. 2012, 287, 40722–40731. [Google Scholar] [PubMed]
- Duckers, H.J.; Boehm, M.; True, A.L.; Yet, S.F.; San, H.; Park, J.L.; Clinton Webb, R.; Lee, M.E.; Nabel, G.J.; Nabel, E.G. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 2001, 7, 693–698. [Google Scholar] [PubMed]
- Li, T.; Tian, H.; Zhao, Y.; An, F.; Zhang, L.; Zhang, J.; Peng, J.; Zhang, Y.; Guo, Y. Heme oxygenase-1 inhibits progression and destabilization of vulnerable plaques in a rabbit model of atherosclerosis. Eur. J. Pharmacol. 2011, 672, 143–152. [Google Scholar] [PubMed]
- Kim, K.M.; Pae, H.O.; Zheng, M.; Park, R.; Kim, Y.M.; Chung, H.T. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase r-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [PubMed]
- Vijayan, V.; Wagener, F.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [PubMed]
- Orozco, L.D.; Kapturczak, M.H.; Barajas, B.; Wang, X.; Weinstein, M.M.; Wong, J.; Deshane, J.; Bolisetty, S.; Shaposhnik, Z.; Shih, D.M.; et al. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ. Res. 2007, 100, 1703–1711. [Google Scholar]
- Ruotsalainen, A.K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Makinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkila, J.; Vatanen, T.; et al. The absence of macrophage nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [PubMed]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. Nf-e2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from lps induced inflammatory response and mortality by cddo-imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar]
- Wang, W.W.; Smith, D.L.; Zucker, S.D. Bilirubin inhibits inos expression and no production in response to endotoxin in rats. Hepatology 2004, 40, 424–433. [Google Scholar]
- Srisook, K.; Han, S.S.; Choi, H.S.; Li, M.H.; Ueda, H.; Kim, C.; Cha, Y.N. Co from enhanced ho activity or from corm-2 inhibits both o2- and no production and downregulates ho-1 expression in lps-stimulated macrophages. Biochem. Pharmacol. 2006, 71, 307–318. [Google Scholar] [PubMed]
- Sharma, H.S.; Maulik, N.; Gho, B.C.; Das, D.K.; Verdouw, P.D. Coordinated expression of heme oxygenase-1 and ubiquitin in the porcine heart subjected to ischemia and reperfusion. Mol. Cell Biochem. 1996, 157, 111–116. [Google Scholar] [PubMed]
- Yet, S.F.; Perrella, M.A.; Layne, M.D.; Hsieh, C.M.; Maemura, K.; Kobzik, L.; Wiesel, P.; Christou, H.; Kourembanas, S.; Lee, M.E. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Investig. 1999, 103, R23–R29. [Google Scholar]
- Liu, X.; Wei, J.; Peng, D.H.; Layne, M.D.; Yet, S.F. Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes 2005, 54, 778–784. [Google Scholar] [PubMed]
- Yet, S.F.; Tian, R.; Layne, M.D.; Wang, Z.Y.; Maemura, K.; Solovyeva, M.; Ith, B.; Melo, L.G.; Zhang, L.; Ingwall, J.S.; et al. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ. Res. 2001, 89, 168–173. [Google Scholar] [PubMed]
- Melo, L.G.; Agrawal, R.; Zhang, L.; Rezvani, M.; Mangi, A.A.; Ehsan, A.; Griese, D.P.; Dell’Acqua, G.; Mann, M.J.; Oyama, J.; et al. Gene therapy strategy for long-term myocardial protection using adeno-associated virus-mediated delivery of heme oxygenase gene. Circulation 2002, 105, 602–607. [Google Scholar] [PubMed]
- Tang, Y.L.; Tang, Y.; Zhang, Y.C.; Qian, K.; Shen, L.; Phillips, M.I. Protection from ischemic heart injury by a vigilant heme oxygenase-1 plasmid system. Hypertension 2004, 43, 746–751. [Google Scholar]
- Tang, Y.L.; Qian, K.; Zhang, Y.C.; Shen, L.; Phillips, M.I. A vigilant, hypoxia-regulated heme oxygenase-1 gene vector in the heart limits cardiac injury after ischemia-reperfusion in vivo. J. Cardiovasc. Pharmacol. Ther. 2005, 10, 251–263. [Google Scholar] [PubMed]
- Issan, Y.; Kornowski, R.; Aravot, D.; Shainberg, A.; Laniado-Schwartzman, M.; Sodhi, K.; Abraham, N.G.; Hochhauser, E. Heme oxygenase-1 induction improves cardiac function following myocardial ischemia by reducing oxidative stress. PLoS ONE 2014, 9, e92246. [Google Scholar]
- Wang, G.; Hamid, T.; Keith, R.J.; Zhou, G.; Partridge, C.R.; Xiang, X.; Kingery, J.R.; Lewis, R.K.; Li, Q.; Rokosh, D.G.; et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 2010, 121, 1912–1925. [Google Scholar]
- Allwood, M.A.; Kinobe, R.T.; Ballantyne, L.; Romanova, N.; Melo, L.G.; Ward, C.A.; Brunt, K.R.; Simpson, J.A. Heme oxygenase-1 overexpression exacerbates heart failure with aging and pressure overload but is protective against isoproterenol-induced cardiomyopathy in mice. Cardiovasc. Pathol. 2014, 23, 231–237. [Google Scholar] [PubMed]
- Bao, W.; Song, F.; Li, X.; Rong, S.; Yang, W.; Zhang, M.; Yao, P.; Hao, L.; Yang, N.; Hu, F.B.; et al. Plasma heme oxygenase-1 concentration is elevated in individuals with type 2 diabetes mellitus. PLoS ONE 2010, 5, e12371. [Google Scholar]
- Sato, T.; Takeno, M.; Honma, K.; Yamauchi, H.; Saito, Y.; Sasaki, T.; Morikubo, H.; Nagashima, Y.; Takagi, S.; Yamanaka, K.; et al. Heme oxygenase-1, a potential biomarker of chronic silicosis, attenuates silica-induced lung injury. Am. J. Respir. Crit. Care Med. 2006, 174, 906–914. [Google Scholar] [PubMed]
- Mateo, I.; Infante, J.; Sanchez-Juan, P.; Garcia-Gorostiaga, I.; Rodriguez-Rodriguez, E.; Vazquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in parkinson’s disease but not in alzheimer’s disease. Acta Neurol. Scand. 2010, 121, 136–138. [Google Scholar] [PubMed]
- Miyazaki, T.; Kirino, Y.; Takeno, M.; Hama, M.; Ushihama, A.; Watanabe, R.; Takase, K.; Tachibana, T.; Matsumoto, K.; Tanaka, M.; et al. Serum ho-1 is useful to make differential diagnosis of secondary hemophagocytic syndrome from other similar hematological conditions. Int. J. Hematol. 2010, 91, 229–237. [Google Scholar] [PubMed]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [PubMed]
- Chen, S.M.; Li, Y.G.; Wang, D.M.; Zhang, G.H.; Tan, C.J. Expression of heme oxygenase-1, hypoxia inducible factor-1alpha, and ubiquitin in peripheral inflammatory cells from patients with coronary heart disease. Clin. Chem. Lab. Med. 2009, 47, 327–333. [Google Scholar]
- Secher, N.; Ostergaard, L.; Tonnesen, E.; Hansen, F.B.; Granfeldt, A. Impact of age on cardiovascular function, inflammation, and oxidative stress in experimental asphyxial cardiac arrest. Acta Anaesthesiol. Scand. 2018, 62, 49–62. [Google Scholar]
- Song, F.; Qi, X.; Chen, W.; Jia, W.; Yao, P.; Nussler, A.K.; Sun, X.; Liu, L. Effect of momordica grosvenori on oxidative stress pathways in renal mitochondria of normal and alloxan-induced diabetic mice. Involvement of heme oxygenase-1. Eur. J. Nutr. 2007, 46, 61–69. [Google Scholar]
- Suzuki, M.; Iso-o, N.; Takeshita, S.; Tsukamoto, K.; Mori, I.; Sato, T.; Ohno, M.; Nagai, R.; Ishizaka, N. Facilitated angiogenesis induced by heme oxygenase-1 gene transfer in a rat model of hindlimb ischemia. Biochem. Biophys. Res. Commun. 2003, 302, 138–143. [Google Scholar]
- Grochot-Przeczek, A.; Kotlinowski, J.; Kozakowska, M.; Starowicz, K.; Jagodzinska, J.; Stachurska, A.; Volger, O.L.; Bukowska-Strakova, K.; Florczyk, U.; Tertil, M.; et al. Heme oxygenase-1 is required for angiogenic function of bone marrow-derived progenitor cells: Role in therapeutic revascularization. Antioxid. Redox Signal. 2014, 20, 1677–1692. [Google Scholar] [PubMed]
- Lee, T.S.; Chang, C.C.; Zhu, Y.; Shyy, J.Y. Simvastatin induces heme oxygenase-1: A novel mechanism of vessel protection. Circulation 2004, 110, 1296–1302. [Google Scholar] [PubMed]
- Heeba, G.; Moselhy, M.E.; Hassan, M.; Khalifa, M.; Gryglewski, R.; Malinski, T. Anti-atherogenic effect of statins: Role of nitric oxide, peroxynitrite and haem oxygenase-1. Br. J. Pharmacol. 2009, 156, 1256–1266. [Google Scholar] [PubMed]
- Wang, Y.; Yu, M.; Ma, Y.; Wang, R.; Liu, W.; Xia, W.; Guan, A.; Xing, C.; Lu, F.; Ji, X. Fenofibrate increases heme oxygenase 1 expression and astrocyte proliferation while limits neuronal injury during intracerebral hemorrhage. Curr. Neurovasc. Res. 2017, 14, 11–18. [Google Scholar] [PubMed]
- Li Volti, G.; Sacerdoti, D.; Di Giacomo, C.; Barcellona, M.L.; Scacco, A.; Murabito, P.; Biondi, A.; Basile, F.; Gazzolo, D.; Abella, R.; et al. Natural heme oxygenase-1 inducers in hepatobiliary function. World J. Gastroenterol. 2008, 14, 6122–6132. [Google Scholar] [PubMed]
- Pittala, V.; Vanella, L.; Salerno, L.; Romeo, G.; Marrazzo, A.; Di Giacomo, C.; Sorrenti, V. Effects of polyphenolic derivatives on heme oxygenase-system in metabolic dysfunctions. Curr. Med. Chem. 2018, 25, 1577–1595. [Google Scholar]
- Pittala, V.; Vanella, L.; Salerno, L.; Di Giacomo, C.; Acquaviva, R.; Raffaele, M.; Romeo, G.; Modica, M.N.; Prezzavento, O.; Sorrenti, V. Novel caffeic acid phenethyl ester (cape) analogues as inducers of heme oxygenase-1. Curr. Pharm Des. 2017, 23, 2657–2664. [Google Scholar] [PubMed]
- Pittala, V.; Salerno, L.; Romeo, G.; Acquaviva, R.; Di Giacomo, C.; Sorrenti, V. Therapeutic potential of caffeic acid phenethyl ester (cape) in diabetes. Curr. Med. Chem. 2018, 25, 4827–4836. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Wang et al., 1998 [14] | Ascending and abdominal aortas | Patients undergoing surgery for CAD (n = 3) or abdominal aortic aneurysm (n = 5) | HO-1 was highly expressed in human atherosclerotic lesions |
| Chen et al., 2005 [15] | Blood leukocytes | Control (n = 30) SAP (n = 30) UAP (n = 40) AMI (n = 35) | HO-1 protein expression was higher in patients with CAD (AMI > UAP > SAP > Control) |
| Ameriso et al., 2005 [16] | Carotid endarterectomy specimens | Controls (n = 7) Patients with symptomatic plaques (n = 25) or asymptomatic plaques (n = 23) | HO-1 expression is highly prevalent in asymptomatic plaques |
| Ijas et al., 2007 [17] | Carotid plaques | (a) Patients with bilateral high-grade stenosis (one being symptomatic and the other asymptomatic) (n = 4) (b) Patients with ipsilateral stroke symptoms (n = 22) or without cerebrovascular symptoms (n = 18) | HO-1 and CD163 were overexpressed in symptomatic carotid plaques in both intra-individual and inter-individual comparison |
| Brydun et al., 2007 [18] | Blood mononuclear cells | 110 patients undergoing coronary angiography | The capacity to upregulate HMOX1 mRNA expression was inversely related to the degree of CAD |
| Cheng et al., 2009 [19] | Carotid endarterectomy specimens | 112 CAD patients | HO-1 protein expression correlated with the vulnerability of atheromatous plaque |
| Idriss et al., 2010 [20] | Plasma | Healthy controls (n = 50) Stable CAD (n = 70) ACS (n = 24) | HO-1 levels were higher in stable CAD and ACS patients |
| Novo et al., 2011 [21] | Serum (or plasma) | Controls (n = 40) AMI (n = 40) | HO-1 levels in AMI patients were significantly higher than in controls, and showed an inverse association with the severity of CAD |
| Yunoki et al., 2013 [22] | Coronary atherectomy specimens | SAP (n = 33) UAP (n = 34) | HO-1-positive areas were significantly higher in UAP patients |
| Li et al., 2014 [23] | Serum | Stroke (n = 60) TIA (n = 50) | HO-1 levels were higher in patients with stroke than TIA |
| Signorelli et al., 2016 [24] | Serum | Controls (n = 27) PAD (n = 27) | HO-1 levels were lower in PAD patients |
| Kishimoto et al., 2018 [25] | Plasma | 136 subjects undergoing carotid ultrasonography for medical check-up | HO-1 levels were high in subjects with carotid plaques |
| Kishimoto et al., 2018 [26] | Plasma | 410 patients undergoing coronary angiography for suspected CAD | HO-1 levels were low in patients with PAD, in contrast to high levels in patients with CAD |
| Fiorelli et al., 2019 [27] | Monocyte-derived macrophages (MDMs) | Healthy controls (10) CAD patients undergoing coronary angiography (30) | HO-1 levels were higher in MDMs of CAD patients and were associated with rupture-prone coronary plaque |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishimoto, Y.; Kondo, K.; Momiyama, Y. The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. Int. J. Mol. Sci. 2019, 20, 3628. https://doi.org/10.3390/ijms20153628
Kishimoto Y, Kondo K, Momiyama Y. The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. International Journal of Molecular Sciences. 2019; 20(15):3628. https://doi.org/10.3390/ijms20153628
Chicago/Turabian StyleKishimoto, Yoshimi, Kazuo Kondo, and Yukihiko Momiyama. 2019. "The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases" International Journal of Molecular Sciences 20, no. 15: 3628. https://doi.org/10.3390/ijms20153628
APA StyleKishimoto, Y., Kondo, K., & Momiyama, Y. (2019). The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. International Journal of Molecular Sciences, 20(15), 3628. https://doi.org/10.3390/ijms20153628