Yeast-Derived β-Glucan in Cancer: Novel Uses of a Traditional Therapeutic

Abstract
1. Introduction
2. The Structure and Signaling of β-Glucan
3. The Trafficking of β-Glucan
4. β-Glucan in Innate and Adaptive Immune Systems
5. β-Glucan and Metabolic Reprogramming
6. Transcriptional Changes Induced by β-Glucan
7. β-Glucan as A Delivery Vesicle for Cancer Therapeutics
8. β-Glucan in Combination Therapy
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCG | Bacillus Calmette-Guerin |
| CR3 | Compliment receptor 3 |
| CR3-DCC | CD3 dependent cellular cytotoxicity |
| DC | Dendritic cells |
| HIF-1α | Hypoxia-inducible factor-1α |
| IP | Intraperitoneal |
| ITAM | immunoreceptor tyrosine-based activation motif |
| IV | Intravenous |
| LLC | Lewis lung carcinoma |
| mAb | Monoclonal antibodies |
| MAC-1 | Macrophage 1 Antigen |
| MDSCs | Myeloid-derived suppressor cells |
| MIF | Migration inhibitory factor |
| M-MDSC | Monocytic MDSC |
| MRD | Minimal residual disease- Minimal residual disease |
| mTOR | mammalian target of Rapamycin |
| ORR | Objective response rate |
| PAMPs | Pathogen-associated molecular patterns |
| PI3K | Phosphatidylinositol-3-kinase |
| PMN-MDSC | Polymorphonuclear MDSC |
| PRR | Pattern recognition receptor |
| TAM | Tumor-associated macrophages- Tumor-associated macrophages |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
| WGP | Whole glucan particles |
References
- Robert, V.A.; Casadevall, A. Vertebrate Endothermy Restricts Most Fungi as Potential Pathogens. J. Infect. Dis. 2009, 200, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Owocki, K.; Kremer, B.; Wrzosek, B.; Królikowska, A.; Kaźmierczak, J. Fungal Ferromanganese Mineralisation in Cretaceous Dinosaur Bones from the Gobi Desert, Mongolia. PLoS ONE 2016, 11, e0146293. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, G.; Sowrirajan, S.; Joseph, B. Extraction and Isolation of beta-Glucan from Grain Sources-A Review. J. Food Sci. 2017, 82, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Ruffing, A.M.; Castro-Melchor, M.; Hu, W.S.; Chen, R.R. Genome sequence of the curdlan-producing Agrobacterium sp. strain ATCC 31749. J. Bacteriol. 2011, 193, 4294–4295. [Google Scholar] [CrossRef] [PubMed]
- Batbayar, S.; Lee, D.H.; Kim, H.W. Immunomodulation of Fungal β-Glucan in Host Defense Signaling by Dectin-1. Biomol. Ther. 2012, 20, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Krizan, J.; Sima, P.; Stakheev, D.; Caja, F.; Rajsiglova, L.; Horak, V.; Saieh, M. Immunostimulatory properties and antitumor activities of glucans (Review). Int. J. Oncol. 2013, 43, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Pillemer, L.; Ecker, E.E. Anticomplementary factor in fresh yeast. J. Biol. Chem. 1941, 137, 139–142. [Google Scholar]
- Riggi, S.J.; Di Luzio, N.R. Identification of a reticuloendothelial stimulating agent in zymosan. Am. J. Physiol. 1961, 200, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Synytsya, A.; Novák, M. Structural diversity of fungal glucans. Carbohydr. Polym. 2013, 92, 792–809. [Google Scholar] [CrossRef]
- Chen, J.; Seviour, R. Medicinal importance of fungal beta-(1->3), (1->6)-glucans. Mycol. Res. 2007, 111, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Yao, J.; Yang, X.; Fang, J. Structural characterization of a water-soluble β-d-glucan from fruiting bodies of Agaricus blazei Murr. Carbohydr. Res. 2002, 337, 1417–1421. [Google Scholar] [CrossRef]
- Ge, Q.; Zhang, A.Q.; Sun, P.L. Isolation, purification and structural characterization of a novel water-soluble glucan from the fruiting bodies of phellinus baumii pilát. J. Food Biochem. 2010, 34, 1205–1215. [Google Scholar] [CrossRef]
- Mandal, S.; Maity, K.K.; Bhunia, S.K.; Dey, B.; Patra, S.; Sikdar, S.R.; Islam, S.S. Chemical analysis of new water-soluble (1→6)-, (1→4)-α, β-glucan and water-insoluble (1→3)-, (1→4)-β-glucan (Calocyban) from alkaline extract of an edible mushroom, Calocybe indica (Dudh Chattu). Carbohydr. Res. 2010, 345, 2657–2663. [Google Scholar] [CrossRef]
- Chakraborty, I.; Mondal, S.; Pramanik, M.; Rout, D.; Islam, S.S. Structural investigation of a water-soluble glucan from an edible mushroom, Astraeus hygrometricus. Carbohydr. Res. 2004, 339, 2249–2254. [Google Scholar] [CrossRef]
- Lowman, D.W.; West, L.J.; Bearden, D.W.; Wempe, M.F.; Power, T.D.; Ensley, H.E.; Haynes, K.; Williams, D.L.; Kruppa, M.D. New insights into the structure of (1->3,1->6)-beta-D-glucan side chains in the Candida glabrata cell wall. PLoS ONE 2011, 6, e27614. [Google Scholar] [CrossRef]
- Chan, G.C.F.; Chan, W.K.; Sze, D.M.Y. The effects of beta-glucan on human immune and cancer cells. J. Hematol. Oncol. 2009, 2, 25. [Google Scholar] [CrossRef]
- Stier, H.; Ebbeskotte, V.; Gruenwald, J. Immune-modulatory effects of dietary Yeast Beta-1,3/1,6-D-glucan. Nutr. J. 2014, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.J.; Kelley, J.L.; Kogan, G.; Ensley, H.E.; Kalbfleisch, J.H.; Browder, I.W.; Williams, D.L. Human monocyte scavenger receptors are pattern recognition receptors for (1->3)-beta-D-glucans. J. Leukoc. Biol. 2002, 72, 140–146. [Google Scholar] [PubMed]
- Zimmerman, J.W.; Lindermuth, J.; Fish, P.A.; Palace, G.P.; Stevenson, T.T.; DeMong, D.E. A novel carbohydrate-glycosphingolipid interaction between a beta-(1-3)-glucan immunomodulator, PGG-glucan, and lactosylceramide of human leukocytes. J. Biol. Chem. 1998, 273, 22014–22020. [Google Scholar] [CrossRef] [PubMed]
- Cain, J.A.; Newman, S.L.; Ross, G.D. Role of complement receptor type three and serum opsonins in the neutrophil response to yeast. Complement 1987, 4, 75–86. [Google Scholar] [CrossRef]
- Ariizumi, K.; Shen, G.L.; Shikano, S.; Xu, S.; Ritter, R.; Kumamoto, T.; Edelbaum, D.; Morita, A.; Bergstresser, P.R.; Takashima, A. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 2000, 275, 20157–20167. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Brown, G.D.; Reid, D.M.; Willment, J.A.; Martinez-Pomares, L.; Gordon, S.; Wong, S.Y. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J. Immunol. 2002, 169, 3876–3882. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006, 6, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.M.; Gow, N.A.; Brown, G.D. Pattern recognition: Recent insights from Dectin-1. Curr. Opin. Immunol. 2009, 21, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Herre, J.; Williams, D.L.; Willment, J.A.; Marshall, A.S.; Gordon, S. Dectin-1 mediates the biological effects of beta-glucans. J. Exp. Med. 2003, 197, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Herre, J.; Marshall, A.S.; Caron, E.; Edwards, A.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.; Reise Sousa, C.; Gordon, S.; Brown, G.D. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004, 104, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Camilli, G.; Tabouret, G.; Quintin, J. The Complexity of Fungal β-Glucan in Health and Disease: Effects on the Mononuclear Phagocyte System. Front. Immunol. 2018, 9, 673. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, X.M.; Heflin, K.E.; Lavigne, L.M.; Yu, K.; Kim, M.; Salomon, A.R.; Reichner, J.S. Lectin Site Ligation of CR3 Induces Conformational Changes and Signaling. J. Biol. Chem. 2012, 287, 3337–3348. [Google Scholar] [CrossRef] [PubMed]
- Vetvicka, V.; Thornton, B.P.; Ross, G.D. Soluble beta-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J. Clin. Invest. 1996, 98, 50–61. [Google Scholar] [CrossRef]
- Vetvicka, V.; Vetvickova, J. 1, 3-Glucan: Silver Bullet or Hot Air? Open Glycosci. 2010, 3, 1–6. [Google Scholar]
- Sletmoen, M.; Stokke, B.T. Higher order structure of (1,3)-beta-D-glucans and its influence on their biological activities and complexation abilities. Biopolymers 2008, 89, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. Fungal beta-glucans and mammalian immunity. Immunity 2003, 19, 311–315. [Google Scholar] [CrossRef]
- Volman, J.J.; Ramakers, J.D.; Plat, J. Dietary modulation of immune function by beta-glucans. Physiol. Behav. 2008, 94, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, I.; Hashimoto, K.; Ohno, N.; Tanaka, H.; Yadomae, T. Immunomodulation by orally administered beta-glucan in mice. Int. J. Immunopharmacol. 1989, 11, 761–769. [Google Scholar] [CrossRef]
- De Graaff, P.; Govers, C.; Wichers, H.J.; Debets, R. Consumption of beta-glucans to spice up T cell treatment of tumors: A review. Expert Opin. Biol. Ther. 2018, 18, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cai, Y.; Qi, C.; Hansen, R.; Ding, C.; Mitchell, T.C.; Yan, J. Orally administered particulate beta-glucan modulates tumor-capturing dendritic cells and improves antitumor T-cell responses in cancer. Clin. Cancer Res. 2010, 16, 5153–5164. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Ross, G.D. Generation of recombinant fragments of CD11b expressing the functional beta-glucan-binding lectin site of CR3 (CD11b/CD18). J. Immunol. 1999, 162, 7285–7293. [Google Scholar] [PubMed]
- Hong, F.; Yan, J.; Baran, J.T.; Allendorf, D.J.; Hansen, R.D.; Ostroff, G.R.; Xing, P.X.; Cheung, N.K.; Ross, G.D. Mechanism by which orally administered β-1, 3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J. Immunol. 2004, 173, 797–806. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef]
- Suzuki, I.; Tanaka, H.; Kinoshita, A.; Oikawa, S.; Osawa, M.; Yadomae, T. Effect of orally administered beta-glucan on macrophage function in mice. Int. J. Immunopharmacol. 1990, 12, 675–684. [Google Scholar] [CrossRef]
- Małaczewska, J.; Wojcik, R.; Jung, L.; Siwicki, A. The effect of Biolex Beta-HP on the selected parameters of biochemical, specific and non-specific humoral and cellular immunity in rats. Bull. Vet. Inst. Pulawy 2010, 54, 75–80. [Google Scholar]
- Leentjens, J.; Quintin, J.; Gerretsen, J.; Kox, M.; Pickkers, P.; Netea, M.G. The Effects of Orally Administered Beta-Glucan on Innate Immune Responses in Humans, a Randomized Open-Label Intervention Pilot-Study. PLoS ONE 2014, 9, e108794. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Allendorf, D.J.; Hansen, R.; Marroquin, J.; Ding, C.; Cramer, D.E.; Yan, J. Yeast β-Glucan Amplifies Phagocyte Killing of iC3b-Opsonized Tumor Cells via Complement Receptor 3-Syk-Phosphatidylinositol 3-Kinase Pathway. J. Immunol. 2006, 177, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Akramiene, D.; Kondrotas, A.; Didziapetriene, J.; Kevelaitis, E. Effects of beta-glucans on the immune system. Medicina 2007, 43, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Babineau, T.J.; Hackford, A.; Kenler, A.; Bistrian, B.; Forse, R.A.; Fairchild, P.G.; Heard, S.; Keroack, M.; Caushaj, P.; Benotti, P. A phase II multicenter, double-blind, randomized, placebo-controlled study of three dosages of an immunomodulator (PGG-glucan) in high-risk surgical patients. Arch. Surg. 1994, 129, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- De Felippe Junior, J.; da Rocha e Silva Junior, M.; Maciel, F.M.; Soares Ade, M.; Mendes, N.F. Infection prevention in patients with severe multiple trauma with the immunomodulator beta 1–3 polyglucose (glucan). Surg. Gynecol. Obstet. 1993, 177, 383–388. [Google Scholar] [PubMed]
- Babineau, T.J.; Marcello, P.; Swails, W.; Kenler, A.; Bistrian, B.; Forse, R.A. Randomized phase I/II trial of a macrophage-specific immunomodulator (PGG-glucan) in high-risk surgical patients. Ann. Surg. 1994, 220, 601–609. [Google Scholar] [CrossRef]
- Vetvicka, V.; Vetvickova, J. A Comparison of Injected and Orally Administered β-glucans. J. Am. Nutraceutical Assoc. 2008, 11, 42–49. [Google Scholar]
- Zheng, X.; Fuling, Z.; Xu, X.; Zhang, L. Uptake of intraperitoneally administrated triple helical β-glucan for antitumor in murine tumor models. J. Mater. Chem. B 2017, 5, 9337–9345. [Google Scholar] [CrossRef]
- Zheng, X.; Lu, F.; Xu, X.; Zhang, L. Extended chain conformation of β-glucan and its effect on antitumor activity. J. Mater. Chem. B 2017, 5, 5623–5631. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance; The Company of Biologists Ltd.: Cambridge, UK, 2012. [Google Scholar]
- Devaud, C.; John, L.B.; Westwood, J.A.; Darcy, P.K.; Kershaw, M.H.J.O. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2013, 2, e25961. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.J.O. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Fluck, M.F.Z.; Rico, M.J.; Matar, P.; Rabinovich, G.A.; Scharovsky, O.G. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol. Immunother. 2007, 56, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peralta, S.; Moraes, C.T. Mitochondrial alterations during carcinogenesis: A review of metabolic transformation and targets for anticancer treatments. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2013; Volume 119, pp. 127–160. [Google Scholar]
- Zhang, M.; Kim, J.A.; Huang, A.Y. Optimizing Tumor Microenvironment for Cancer Immunotherapy: β-Glucan-Based Nanoparticles. Front. Immunol. 2018, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Canavera, S.J.; Akira, S.; Underhill, D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003, 197, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Cai, Y.; Gunn, L.; Ding, C.; Li, B.; Kloecker, G.; Qian, K.; Vasilakos, J.; Saijo, S.; Iwakura, Y.J.B. Differential pathways regulating innate and adaptive antitumor immune responses by particulate and soluble yeast-derived β-glucans. Blood 2011, 117, 6825–6836. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Větvička, V.; Yan, J.; Hanikýřová, M.; Mayadas, T.; Ross, G.D. The β-glucan-binding lectin site of mouse CR3 (CD11b/CD18) and its function in generating a primed state of the receptor that mediates cytotoxic activation in response to iC3b-opsonized target cells. J. Immunol. 1999, 162, 2281–2290. [Google Scholar] [PubMed]
- Wu, T.C.; Xu, K.; Banchereau, R.; Marches, F.; Chun, I.Y.; Martinek, J.; Anguiano, E.; Pedroza-Gonzalez, A.; Snipes, G.J.; O’Shaughnessy, J. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol. Res. 2014, 2, 487–500. [Google Scholar] [CrossRef]
- Osorio, F.; LeibundGut-Landmann, S.; Lochner, M.; Lahl, K.; Sparwasser, T.; Eberl, G.; Reis e Sousa, C. DC activated via dectin-1 convert Treg into IL-17 producers. Eur. J. Immunol. 2008, 38, 3274–3281. [Google Scholar] [CrossRef]
- Mantovani, A.J.N. Cancer: Inflaming metastasis. Nature 2009, 457, 36. [Google Scholar] [CrossRef]
- Campbell, M.J.; Tonlaar, N.Y.; Garwood, E.R.; Huo, D.; Moore, D.H.; Khramtsov, A.I.; Au, A.; Baehner, F.; Chen, Y.; Malaka, D.O.; et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2011, 128, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.T.; Lee, K.Y.; Wang, C.W.; Heh, C.C.; Chan, Y.F.; Chen, H.W.; Kuo, C.H.; Feng, P.H.; Lin, T.Y.; Wang, C.H. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int. J. Cancer 2012, 131, E227–E235. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Luo, F.; Ding, C.; Albeituni, S.; Hu, X.; Ma, Y.; Cai, Y.; McNally, L.; Sanders, M.A.; Jain, D. Dectin-1 activation by a natural product β-glucan converts immunosuppressive macrophages into an M1-like phenotype. J. Immunol. 2015, 195, 5055–5065. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.H.; Ding, C.; Liu, M.; Hu, X.; Luo, F.; Kloecker, G.; Bousamra, M.; Zhang, H.G.; Yan, J. Yeast-derived particulate β-glucan treatment subverts the suppression of myeloid-derived suppressor cells (MDSC) by inducing polymorphonuclear MDSC apoptosis and monocytic MDSC differentiation to APC in cancer. J. Immunol. 2016, 196, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.F.; Driscoll, C.B.; Walters, P.R.; Limper, A.H.; Carmona, E.M. β-glucan–activated human B lymphocytes participate in innate immune responses by releasing proinflammatory cytokines and stimulating neutrophil chemotaxis. J. Immunol. 2015, 195, 5318–5326. [Google Scholar] [CrossRef] [PubMed]
- Harnack, U.; Kellermann, U.; Pecher, G. Yeast-derived beta-(1-3),(1-6)-D-glucan induces up-regulation of CD86 on dectin-1-positive human B-lymphoma cell lines. Anticancer Res. 2011, 31, 4195–4199. [Google Scholar] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889. [Google Scholar] [CrossRef]
- Puig-Kroger, A.; Sierra-Filardi, E.; Dominguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gomez-Aguado, F.; Ratnam, M.; Sanchez-Mateos, P.; Corbi, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR-and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed]
- Mourits, V.P.; Wijkmans, J.C.H.M.; Joosten, L.A.B.; Netea, M.G. Trained immunity as a novel therapeutic strategy. Curr. Opin. Pharmacol. 2018, 41, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Moorlag, S.; Novakovic, B.; Li, Y.; Wang, S.Y.; Oosting, M.; Kumar, V.; Xavier, R.J.; Wijmenga, C.; Joosten, L.A.B.; et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 2018, 23, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.W.M.; Joosten, L.A.B.; Riksen, N.; Netea, M.G. Trained immunity: A smart way to enhance innate immune defence. Mol. Immunol. 2015, 68, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans Infection Affords Protection against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368. [Google Scholar] [CrossRef]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146. [Google Scholar] [CrossRef]
- Buffen, K.; Oosting, M.; Quintin, J.; Ng, A.; Kleinnijenhuis, J.; Kumar, V.; van de Vosse, E.; Wijmenga, C.; van Crevel, R.; Oosterwijk, E.; et al. Autophagy Controls BCG-Induced Trained Immunity and the Response to Intravesical BCG Therapy for Bladder Cancer. PLoS Pathog. 2014, 10, e1004485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Choksi, S.; Liu, Z. Chapter 12—Autophagy Is Required During Monocyte–Macrophage Differentiation. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 195–206. [Google Scholar]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Lefebvre, M.L.; Labroquere, K.; Faure, O.; Abastado, J.P. Liposomal muramyl tripeptide phosphatidylethanolamine: Targeting and activating macrophages for adjuvant treatment of osteosarcoma. Curr. Cancer Drug Targets 2006, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Eidinger, D.; Bruce, A.W. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J. Urol. 1976, 116, 180–183. [Google Scholar] [CrossRef]
- Luo, Y.; Knudson, M.J. Mycobacterium bovis bacillus Calmette-Guerin-induced macrophage cytotoxicity against bladder cancer cells. Clin. Dev. Immunol. 2010, 2010, 357591. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Miyazaki, J.; Joraku, A.; Nishiyama, H.; Akaza, H. Bacillus Calmette-Guerin (BCG) immunotherapy for bladder cancer: Current understanding and perspectives on engineered BCG vaccine. Cancer Sci. 2013, 104, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.P.; Fiering, S.N.; Ostroff, G.R.; Cramer, R.A.; Mullins, D.W. Beta-glucan-induced trained innate immunity mediates antitumor efficacy in the mouse lung. J. Immunol. 2016, 196, 1731–1742. [Google Scholar]
- Alexander, M.P.; Fiering, S.N. Beta-glucan-induced inflammatory monocytes mediate antitumor efficacy in the murine lung. Cancer Immunol. Immunother. 2018, 67, 1731–1742. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; van der Meer, J.W.M. Hypothesis: Stimulation of trained immunity as adjunctive immunotherapy in cancer. J. Leukoc. Biol. 2017, 102, 1323–1332. [Google Scholar] [CrossRef]
- Sancho, D.; Reis e Sousa, C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu. Rev. Immunol. 2012, 30, 491–529. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M.J.C. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M.J.C. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Connelly, L.; Barham, W.; Onishko, H.M.; Chen, L.; Sherrill, T.P.; Zabuawala, T.; Ostrowski, M.C.; Blackwell, T.S.; Yull, F.E. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011, 13, R83. [Google Scholar] [CrossRef] [PubMed]
- Elder, M.J.; Webster, S.J.; Chee, R.; Williams, D.L.; Hill Gaston, J.S.; Goodall, J.C. β-Glucan Size Controls Dectin-1-Mediated Immune Responses in Human Dendritic Cells by Regulating IL-1β Production. Front. Immunol. 2017, 8, 791. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. The macrophage: Past, present and future. Eur. J. Immunol. 2007, 37 (Suppl. 1), S9–S17. [Google Scholar] [CrossRef]
- Zhang, X.; Edwards, J.P.; Mosser, D.M. The expression of exogenous genes in macrophages: Obstacles and opportunities. Methods Mol. Biol. 2009, 531, 123–143. [Google Scholar] [CrossRef]
- Basha, G.; Novobrantseva, T.I.; Rosin, N.; Tam, Y.Y.; Hafez, I.M.; Wong, M.K.; Sugo, T.; Ruda, V.M.; Qin, J.; Klebanov, B.; et al. Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 2186–2200. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Zhang, M.; Gao, Y.; Caja, K.; Zhao, B.; Kim, J.A. Non-Viral Nanoparticle Delivers Small Interfering RNA to Macrophages In Vitro and In Vivo. PLoS ONE 2015, 10, e0118472. [Google Scholar] [CrossRef]
- Soto, E.R.; Caras, A.C.; Kut, L.C.; Castle, M.K.; Ostroff, G.R. Glucan Particles for Macrophage Targeted Delivery of Nanoparticles. J. Drug Deliv. 2012, 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.P.; Rinn, B.; Meyer, B.; Dodel, R.; Bacher, M. Role of MIF in Inflammation and Tumorigenesis. Oncology 2008, 75, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, L.; Kim, J.A. Modulating mammary tumor growth, metastasis and immunosuppression by siRNA-induced MIF reduction in tumor microenvironment. Cancer Gene Ther. 2015, 22, 463–474. [Google Scholar] [CrossRef]
- Aouadi, M.; Tesz, G.J.; Nicoloro, S.M.; Wang, M.; Chouinard, M.; Soto, E.; Ostroff, G.R.; Czech, M.P. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 2009, 458, 1180. [Google Scholar] [CrossRef] [PubMed]
- Tesz, G.J.; Aouadi, M.; Prot, M.; Nicoloro, S.M.; Boutet, E.; Amano, S.U.; Goller, A.; Wang, M.; Guo, C.A.; Salomon, W.E.; et al. Glucan particles for selective delivery of siRNA to phagocytic cells in mice. Biochem. J. 2011, 436, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Kaufmann, S.H. How does doxorubicin work? Elife 2012, 1, e00387. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Lee, K.; Gilad, A.; Choi, J. Synthesis of Beta-glucan Nanoparticles for the Delivery of Single Strand DNA. Biotechnol. Bioprocess Eng. 2018, 23, 144–149. [Google Scholar] [CrossRef]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Berner, V.K.; duPre, S.A.; Redelman, D.; Hunter, K.W. Microparticulate β-glucan vaccine conjugates phagocytized by dendritic cells activate both naïve CD4 and CD8 T cells in vitro. Cell. Immunol. 2015, 298, 104–114. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Wang, Y.; Liu, F.; Fernandez-Tejada, A.; Dong, S. beta-Glucan as an immune activator and a carrier in the construction of a synthetic MUC1 vaccine. Chem. Commun. 2018, 55, 253–256. [Google Scholar] [CrossRef]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.K.; Kmieciak, M.; Graham, L.; Feldmesser, M.; Bear, H.D.; Manjili, M.H. Adoptive transfer of HER2/neu-specific T cells expanded with alternating gamma chain cytokines mediate tumor regression when combined with the depletion of myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2009, 58, 941. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Li, P.; Wang, F. β-glucans as potential immunoadjuvants: A review on the adjuvanticity, structure-activity relationship and receptor recognition properties. Vaccine 2018, 36, 5235–5244. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Hansen, R.D.; Yan, J.; Allendorf, D.J.; Baran, J.T.; Ostroff, G.R.; Ross, G.D. β-Glucan functions as an adjuvant for monoclonal antibody immunotherapy by recruiting tumoricidal granulocytes as killer cells. Cancer Res. 2003, 63, 9023–9031. [Google Scholar] [PubMed]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Kramer, K.; Ragupathi, G.; Cheung, N.K. Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin. Cancer Res. 2014, 20, 1375–1382. [Google Scholar] [CrossRef]
- Modak, S.; Kushner, B.H.; Kramer, K.; Vickers, A.; Cheung, I.Y.; Cheung, N.K.V. Anti-GD2 antibody 3F8 and barley-derived (1→3), (1→4)-β-D-glucan: A Phase I study in patients with chemoresistant neuroblastoma. Oncoimmunology 2013, 2, e23402. [Google Scholar] [CrossRef]
- Hirschowitz, E.A.; Mullins, A.; Prajapati, D.; Baeker, T.; Kloecker, G.; Foody, T.; Damron, K.; Love, C.; Yannelli, J.R. Pilot study of 1650-G: A simplified cellular vaccine for lung cancer. J. Thorac. Oncol. 2011, 6, 169–173. [Google Scholar] [CrossRef]
- O’Day, S.; Borges, V.; Chmielowski, B.; Rao, R.; Abu-Khalaf, M.; Stopeck, A.; Lowe, J.; Mattson, P.; Breuer, K.; Gargano, M.; et al. Abstract P2-09-08: Imprime PGG, a novel innate immune modulator, combined with pembrolizumab in a phase 2 multicenter, open label study in chemotherapy-resistant metastatic triple negative breast cancer (TNBC). Cancer Res. 2019, 79, P2–P9. [Google Scholar]
- Fraser, K.; Chan, A.; Fulton, R.; Leonardo, S.; Jonas, A.; Qiu, X.; Ottoson, N.; Kangas, T.; Gordon, K.; Graff, J. Imprime PGG triggers PD-L1 expression on tumor and myeloid cells and prevents tumor establishment in combination with αPD-L1 treatment in vivo. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]
- Qiu, X.; Chan, A.S.; Jonas, A.B.; Kangas, T.; Ottoson, N.R.; Graff, J.R.; Bose, N. Imprime PGG, a yeast β-glucan PAMP elicits a coordinated immune response in combination with anti-PD1 antibody. Am. Assoc. Immnol. 2016, 196, 16. [Google Scholar]
- Weitberg, A.B. A phase I/II trial of beta-(1, 3)/(1, 6) D-glucan in the treatment of patients with advanced malignancies receiving chemotherapy. J. Exp. Clin. Cancer Res. 2008, 27, 40. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Gada, P.; Senzer, N.; Gargano, M.A.; Patchen, M.L.; Saltz, L.B. A phase II efficacy and safety, open-label, multicenter study of imprime PGG injection in combination with cetuximab in patients with stage IV KRAS-mutant colorectal cancer. Clin. Colorectal Cancer 2016, 15, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Sadjadian, P.; Kollmeier, J.; Lowe, J.; Mattson, P.; Trout, J.R.; Gargano, M.; Patchen, M.L.; Walsh, R.; Beliveau, M.; et al. A randomized, open-label, multicenter, phase II study evaluating the efficacy and safety of BTH1677 (1,3-1,6 beta glucan; Imprime PGG) in combination with cetuximab and chemotherapy in patients with advanced non-small cell lung cancer. Investig. New Drugs 2017, 35, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.A.; Redd, R.; Reynolds, C.; Fields, M.; Armand, P.; Fisher, D.C.; Jacobsen, E.D.; LaCasce, A.S.; Bose, N.; Ottoson, N.; et al. A phase 2 clinical trial of rituximab and β-glucan pgg in relapsed/refractory indolent B-cell non-hodgkin lymphoma. Hematol. Oncol. 2019, 37, 521–523. [Google Scholar] [CrossRef]


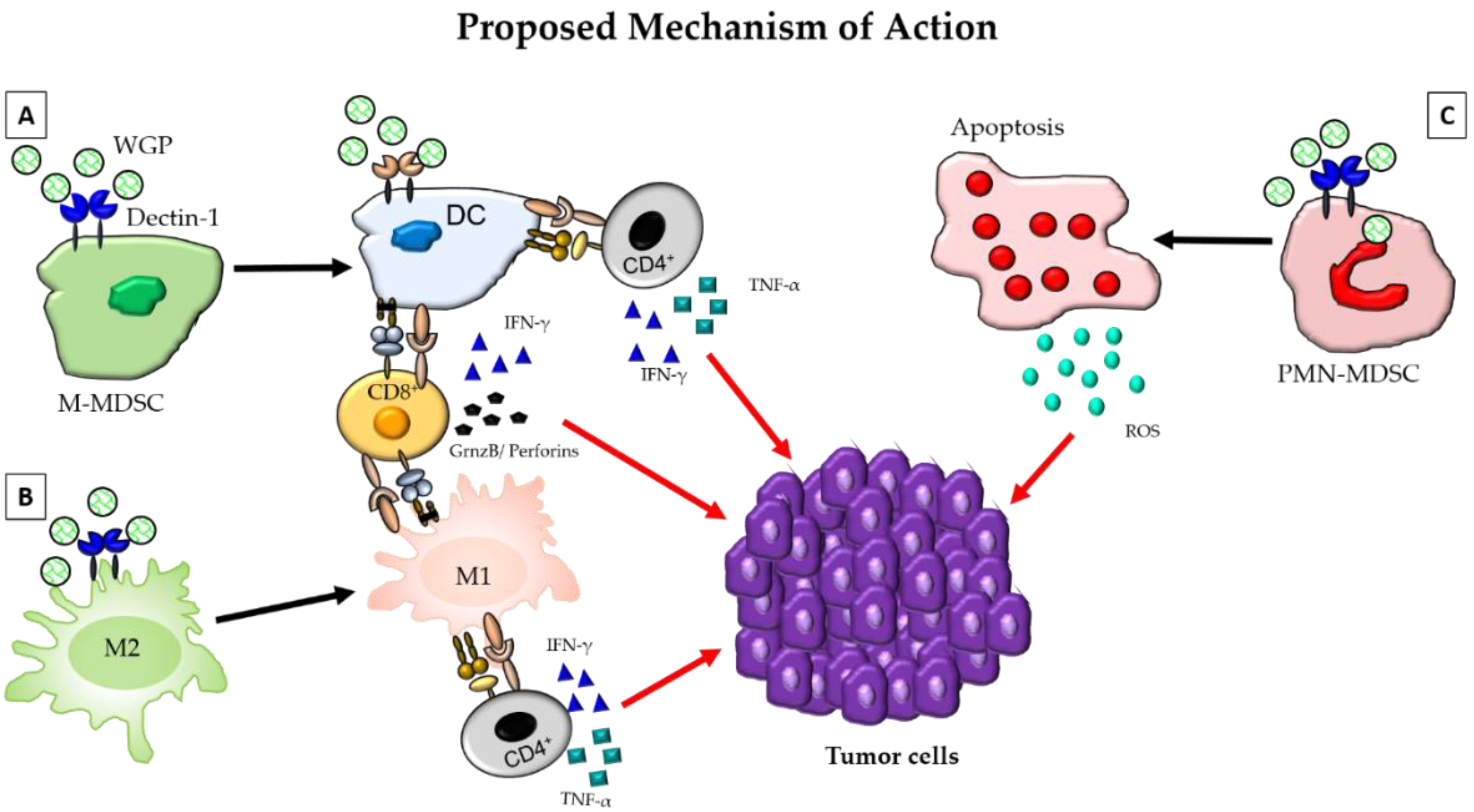{kind=link}
| Therapeutic Delivered | Glucan Utilized | Delivery Mechanism/Mechanism of Action | Results of Study | Reference |
|---|---|---|---|---|
| Anti-MIF siRNA | β-(1,3)(1,4)-D-glucan | A glucan based siRNA carrier system (BG34-10-Re-I) delivered IV that can effectively assemble siRNA into uniformly distributed nanoparticles that are delivered to macrophages. | Injection of the nanoparticles into Balb/c mice bearing 4T1 mammary tumors resulted in MIF reduction in TAMs | [106] |
| Anti-TNFα siRNA | β-(1,3)-D-glucan | β1,3-d-glucan-encapsulated siRNA particles (GeRPs) were used as oral delivery vehicles. After oral consumption, particles were phagocytosed by macrophages and DCs in the GALT, then trafficked to other reticuloendothelial system tissues. | Oral gavage of mice with GeRPs containing as little as 20 µg kg−1 siRNA directed against TNFα depleted its messenger RNA in macrophages recovered from the peritoneum, spleen, liver and lung, and lowered serum TNFα levels. | [107] |
| Doxorubicin | β-(1,3)-D-glucan | Glucan particle nanoparticle formulations were made with fluorescent anionic polystyrene nanoparticles. Mesoporous silica nanoparticles (MSNs) containing doxorubicin were bound to the glucan nanoparticles | Doxorubicin loaded glucan particles effectively delivered doxorubicin into phagocytic ells resulting in enhanced cell-cycle arrest in vitro. | [104] |
| ssDNA | Zymosan | β-glucan nanoparticles (GluNPs) were prepared by slicing β -glucan into low molecular weight using various concentrations of Trifluoroacetic acid (TFA). ssDNA was then inserted into the glucan triple helix structure. | The group successfully produced a low molecular weight β-glucan containing ssDNA. The particles have not yet been used to test therapeutic efficacy. | [111] |
| MUC-1 peptide antigen | β-(1,3)-D-glucan | A Muc-1 peptide antigen was conjugated to β-glucan with the goal of creating a cancer vaccine candidate. It was hypothesized that recognition, and uptake of the conjugate by myeloid cells may activate innate immunity while the presentation of the MUC1 epitope may activate adaptive immunity, leading to overall more robust responses to tumors displaying high levels of MUC-1 | Mice subcutaneously receiving the conjugate showed high anti-MUC1 antibody titers cross-reactive with the MCF-7 tumor cell line, as well as high levels of IL-6 and IFN-γ in sera. Overall the conjugate was able to elicit potent immune responses and immunogenicity of the MUC-1 peptide enhanced through conjugation to β-glucan. | [113] |
| Combination | Type of Cancer | Status | Clinical Trial ID | Outcomes |
|---|---|---|---|---|
| β-Glucan with OPT-821 and vaccine therapy(β-glucan and OPT-821 as immunoadjuvants to modulate the immune system and vaccine to produce an effective immune response against the tumor cells) | Neuroblastoma | Phase I/II | NCT00911560 | No dose-limiting toxicity reported at 150 µg/m2 of OPT-821, relapse-free survival was reported to be 80% ± 10% at 24 months, significant antibody response against GD2 and/or GD3 (OPT-821) in 12 out of 15 patients [120] |
| Pembrolizumab with Imprime PGG (Imprime PGG enhances the sensitivity to checkpoint inhibitors by activating the innate and adaptive immune responses) | Chemotherapy-resistant metastatic triple-negative breast cancer | Phase II | NCT02981303 | 15.9% Overall response rate (ORR); 25% disease control rates for >24 weeks; 13.7 months overall survival with 79% patient survival up to six months, 71.5% survival up to nine months and 64.2% patient survival up to 12 months [123]. |
| Pembrolizumab with Imprime PGG | Metastatic Non-small cell lung cancer | Phase I/II | NCT03003468 | Trial ongoing |
| Monoclonal antibody 3F8 with β–glucan(3F8 blocks tumor growth and β-glucan stimulates the immune system to kill tumor cells) | Metastatic Neuroblastoma | Phase I | NCT00492167 | Combination therapy of 3F8 along with β-glucan has been reported to be well-tolerated with antineoplastic activity [121]. |
| Rituximab and β-glucan(rituximab locates cancer cells enabling cancer cell killing or delivering cancer-killing substances to the cancer site and combination with β-glucan increases the effectiveness of rituximab by making cancer cells more sensitive to the antibody) | Relapsed or Progressive lymphoma or Leukemia, or Transplantation-related Lymphoproliferative Disorder | Phase I | NCT00087009 | Trial Ongoing |
| Rituximab and Imprime PGG(Imprime PGG activates innate immune cells through CR3 and combination with rituximab improves the responses through complement-dependent cytoxicity) | Relapsed Indolent Non-Hodgkin Lymphoma | Phase II | NCT02086175 | Combination of β-glucan with rituximab was well-tolerated with 46% observed response rate, resulted in pro-inflammatory cytokines related to M1-macrophage phenotype, increased antigen presentation and expansion and activation of tumor-infiltrating T cells [129]. |
| Lung cancer vaccine 1650-G and GM-CSF with β–glucan(1650-G is a vaccine comprising killed allogeneic tumor cells that act as tumor antigens, GM-CSF acts as an adjuvant) | Stage I-IIA Non-Small Cell Lung cancer (NSCLC) | Phase I/II | NCT01829373 | Combination treatment is reported to be safe with a robust immunologic response in 6 out of 11 patients, and the kinetics of the responses were observed similar to that with DC vaccines indicating a promising and cheaper multivalent vaccine for NSCLC [122]. |
| Cetuximab/Carboplatin/Paclitaxel in combination with Imprime PGG | Advanced Non-Small Cell Lung Cancer | Phase II | NCT00874848 | The combination treatment was well-tolerated and showed improved overall response rate (ORR) [128]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geller, A.; Shrestha, R.; Yan, J. Yeast-Derived β-Glucan in Cancer: Novel Uses of a Traditional Therapeutic. Int. J. Mol. Sci. 2019, 20, 3618. https://doi.org/10.3390/ijms20153618
Geller A, Shrestha R, Yan J. Yeast-Derived β-Glucan in Cancer: Novel Uses of a Traditional Therapeutic. International Journal of Molecular Sciences. 2019; 20(15):3618. https://doi.org/10.3390/ijms20153618
Chicago/Turabian StyleGeller, Anne, Rejeena Shrestha, and Jun Yan. 2019. "Yeast-Derived β-Glucan in Cancer: Novel Uses of a Traditional Therapeutic" International Journal of Molecular Sciences 20, no. 15: 3618. https://doi.org/10.3390/ijms20153618
APA StyleGeller, A., Shrestha, R., & Yan, J. (2019). Yeast-Derived β-Glucan in Cancer: Novel Uses of a Traditional Therapeutic. International Journal of Molecular Sciences, 20(15), 3618. https://doi.org/10.3390/ijms20153618