Non-Cytotoxic Agarose/Hydroxyapatite Composite Scaffolds for Drug Release

 ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
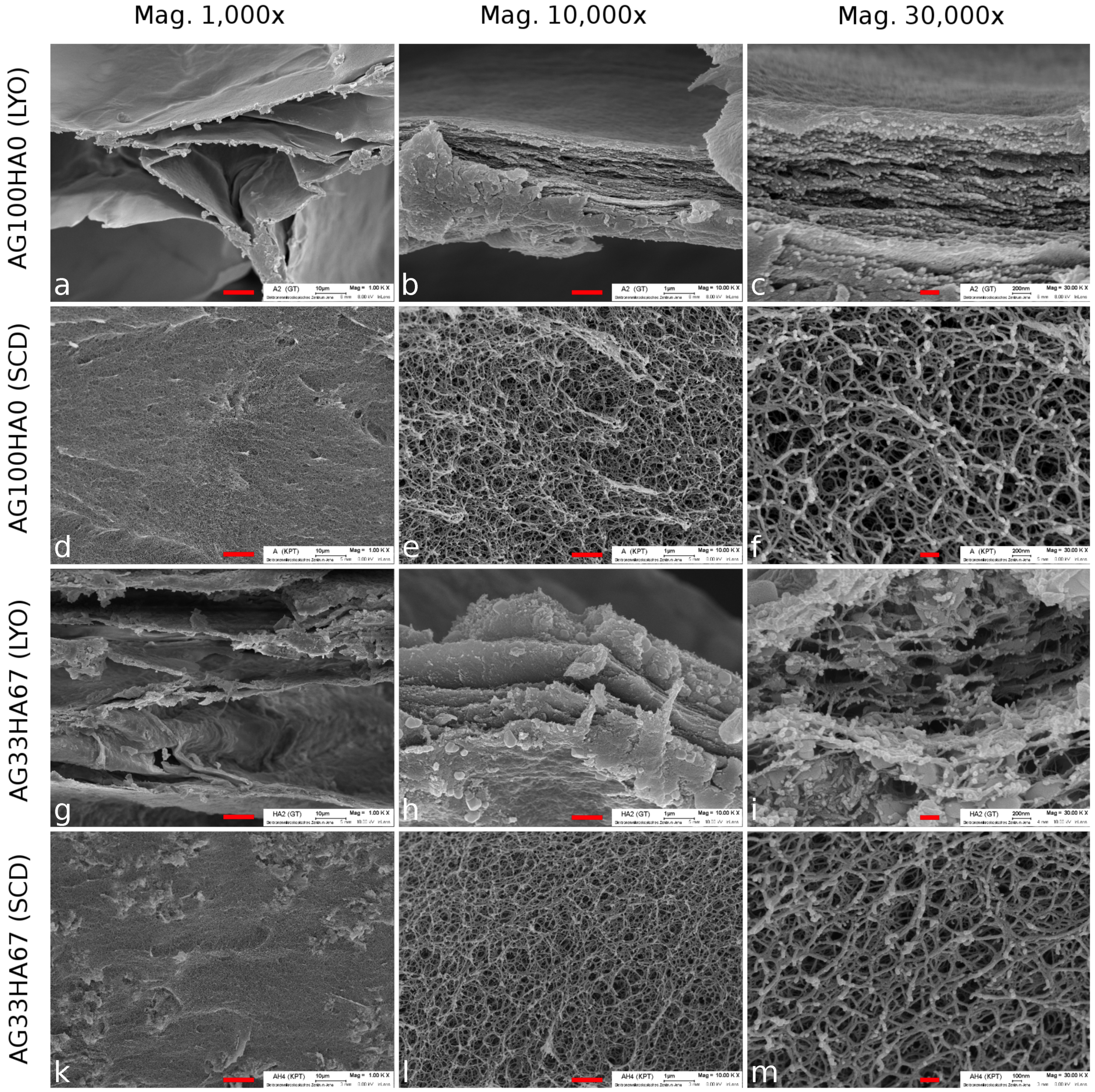
2.1. Scaffold Characterization
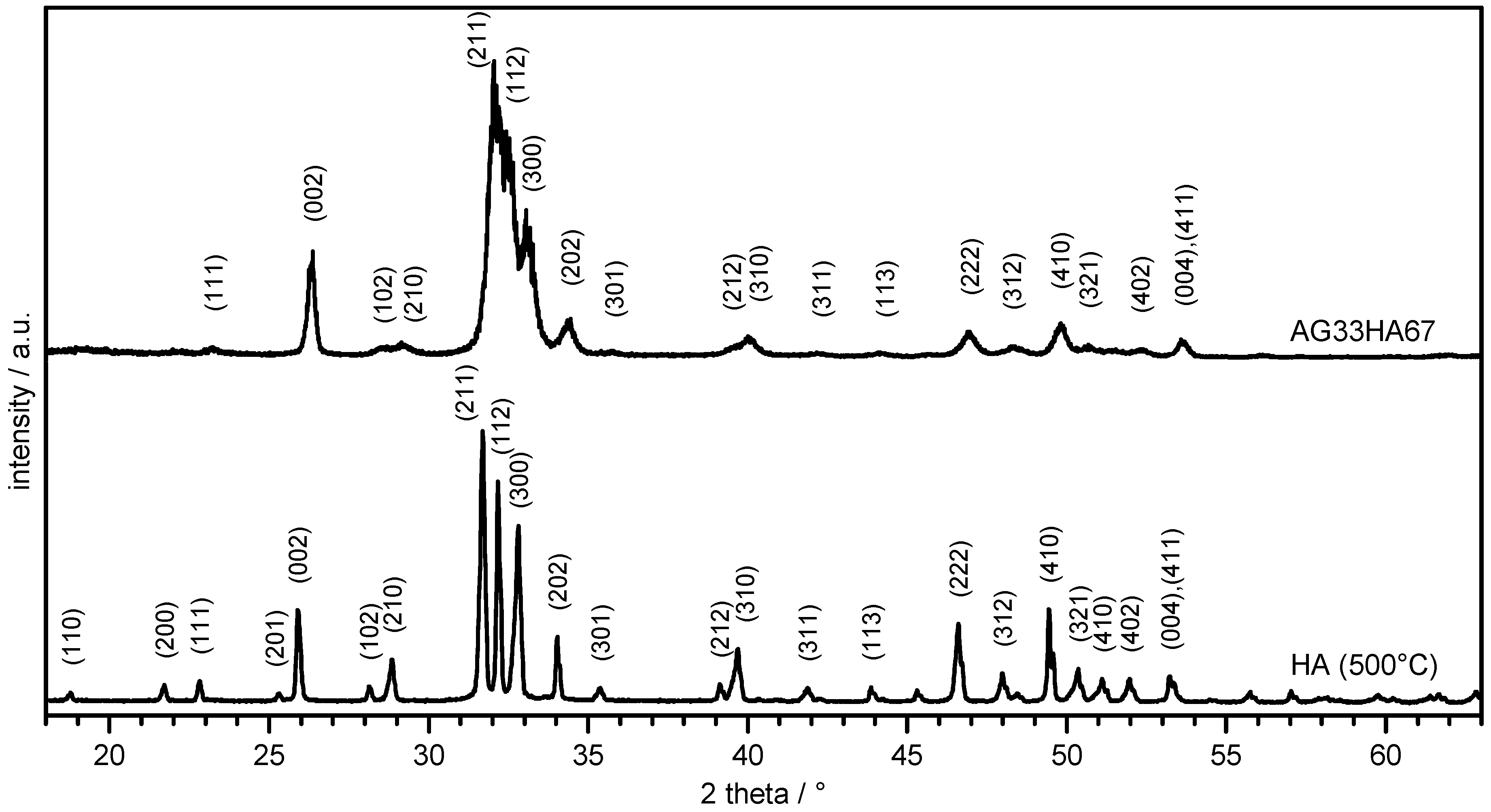
2.1.1. X-Ray Diffraction
2.1.2. Infrared Spectroscopy
2.1.3. Porosity
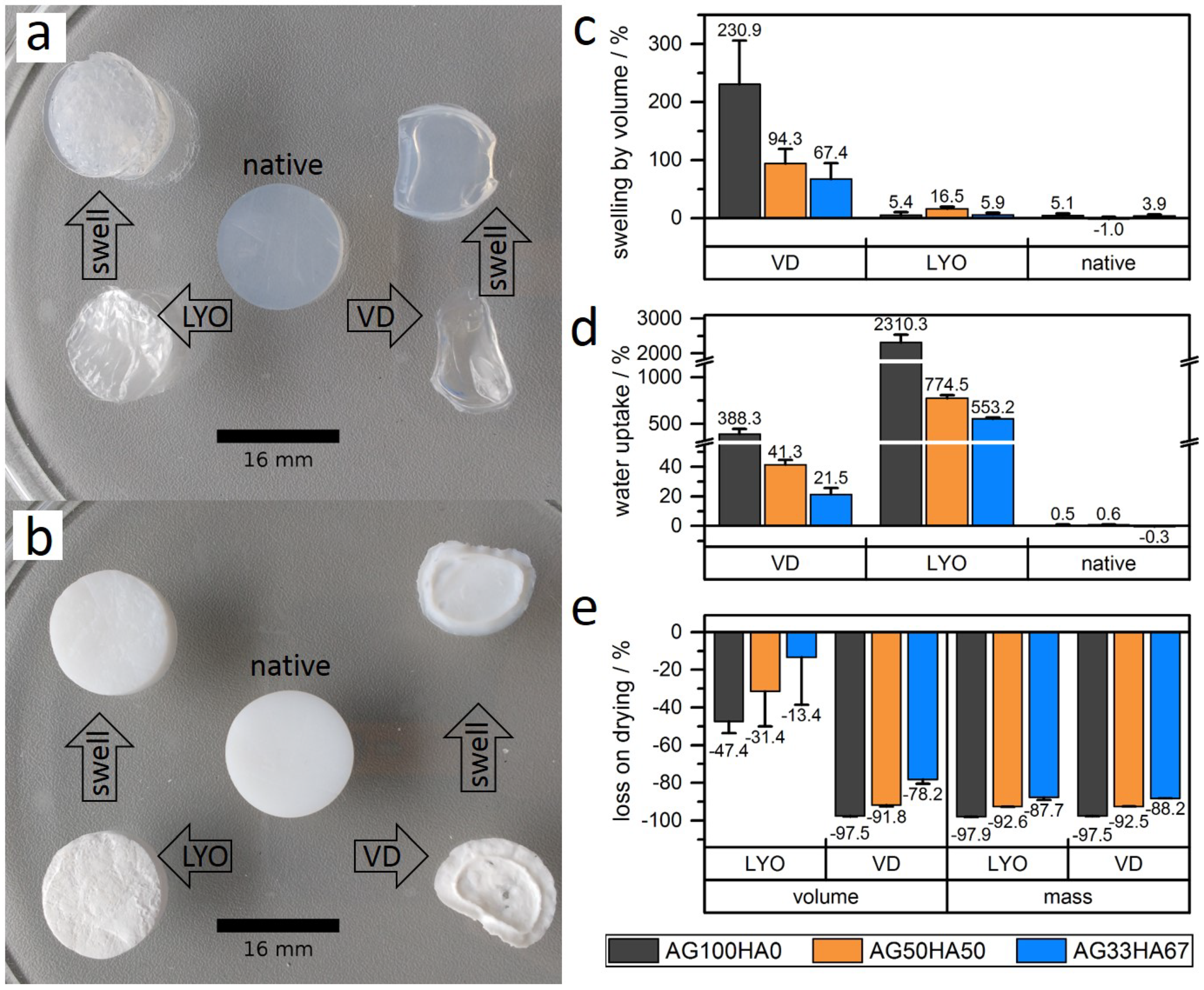
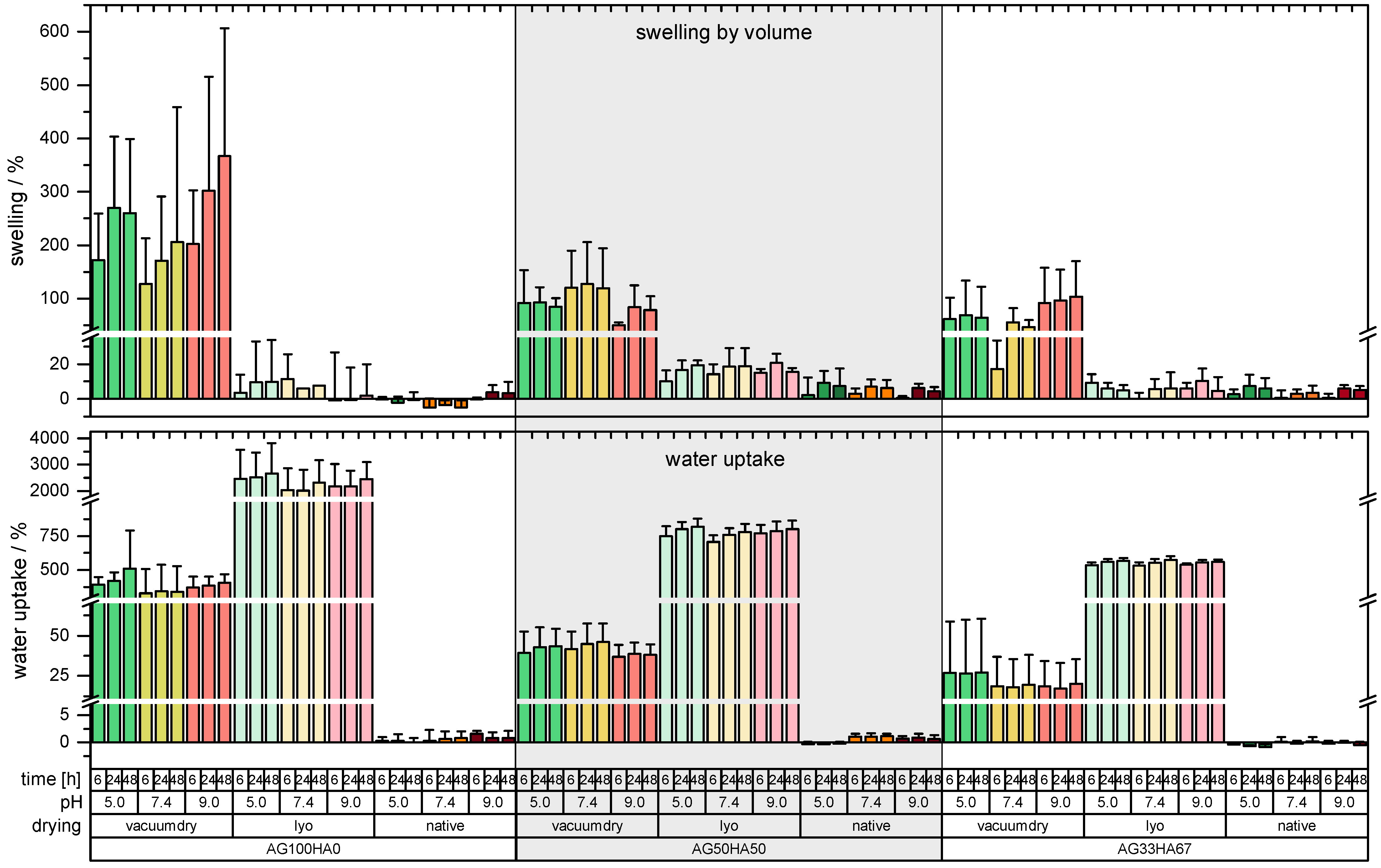
2.1.4. Shrinking and Swelling
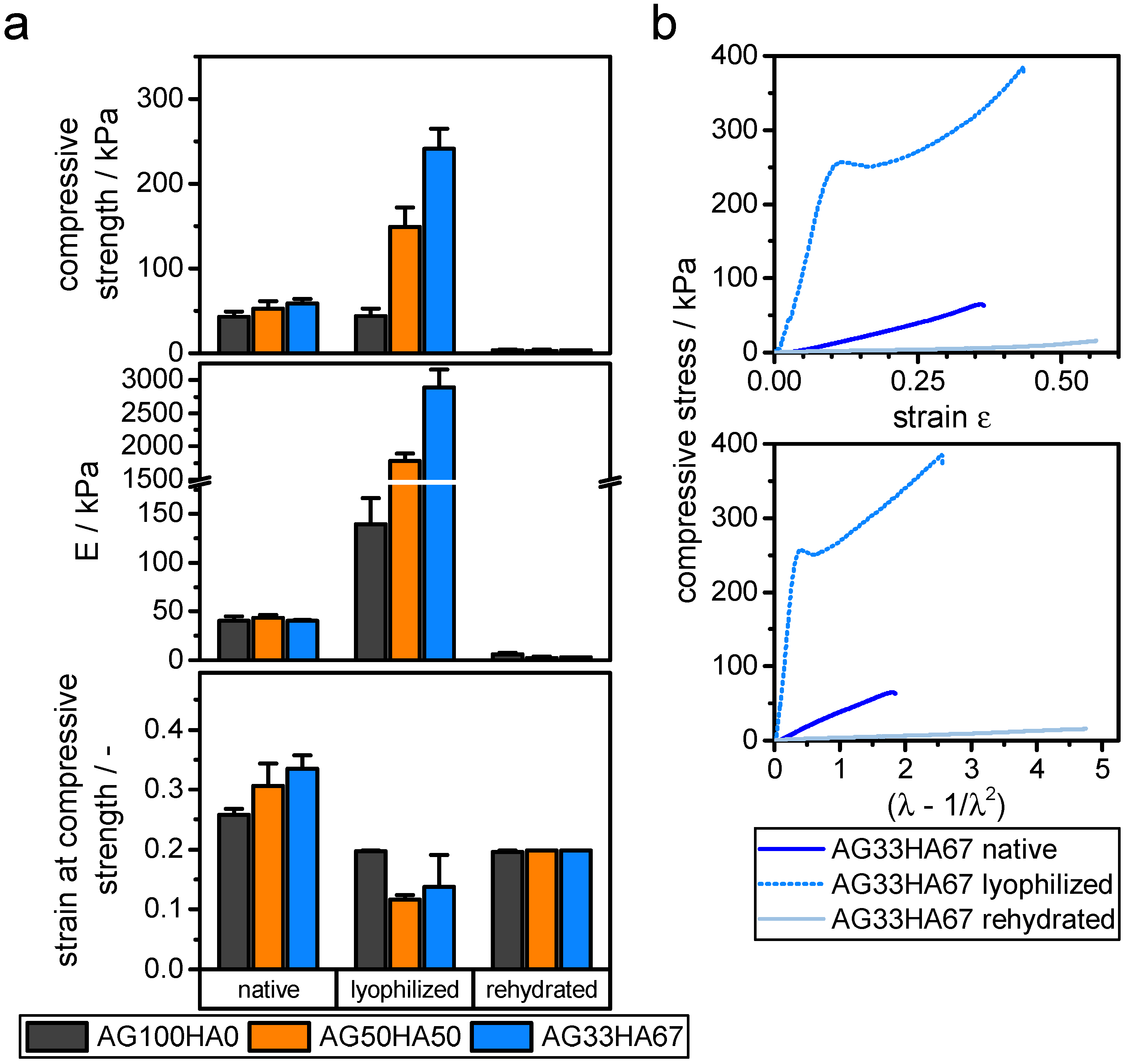
2.1.5. Mechanical Properties
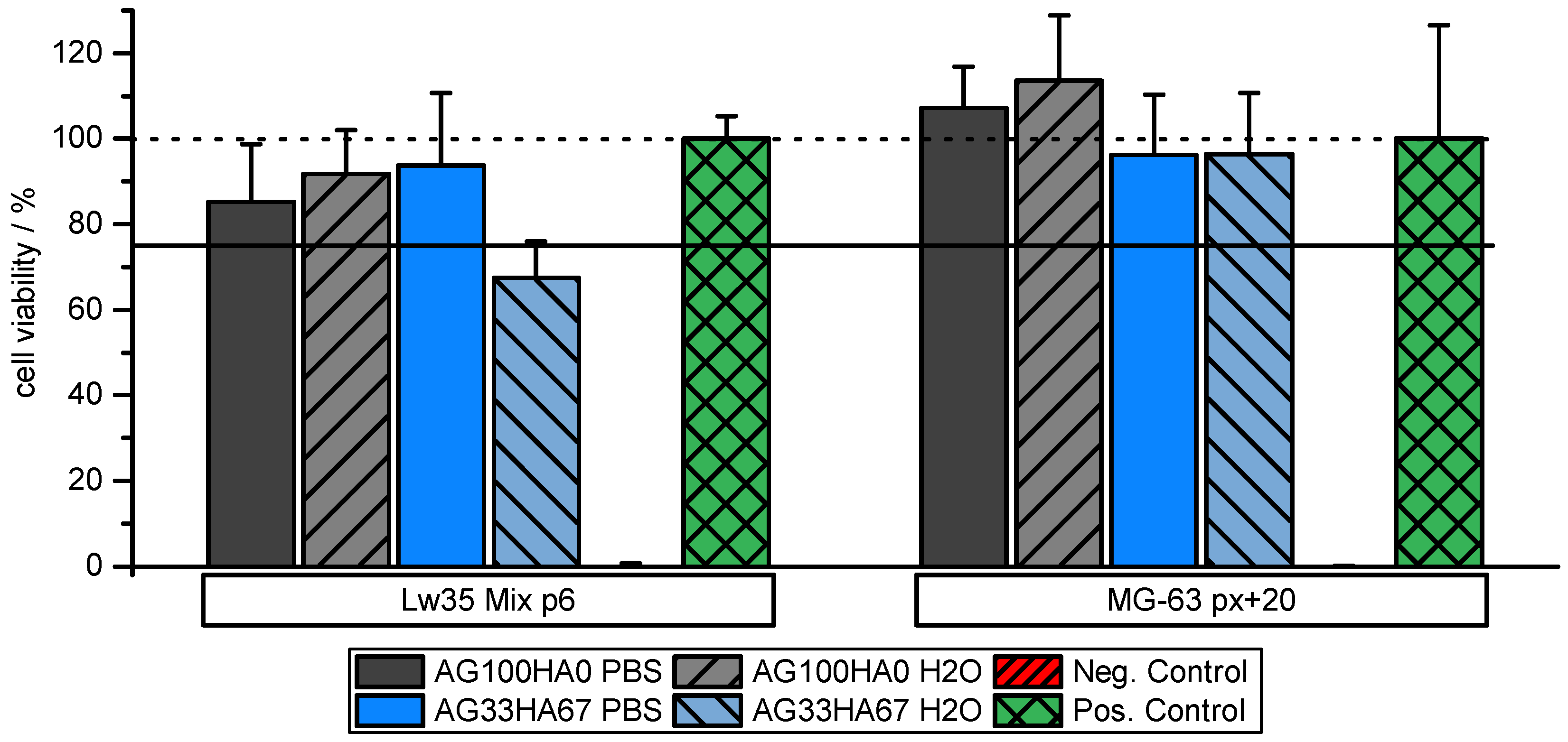
2.2. Biocompatibility
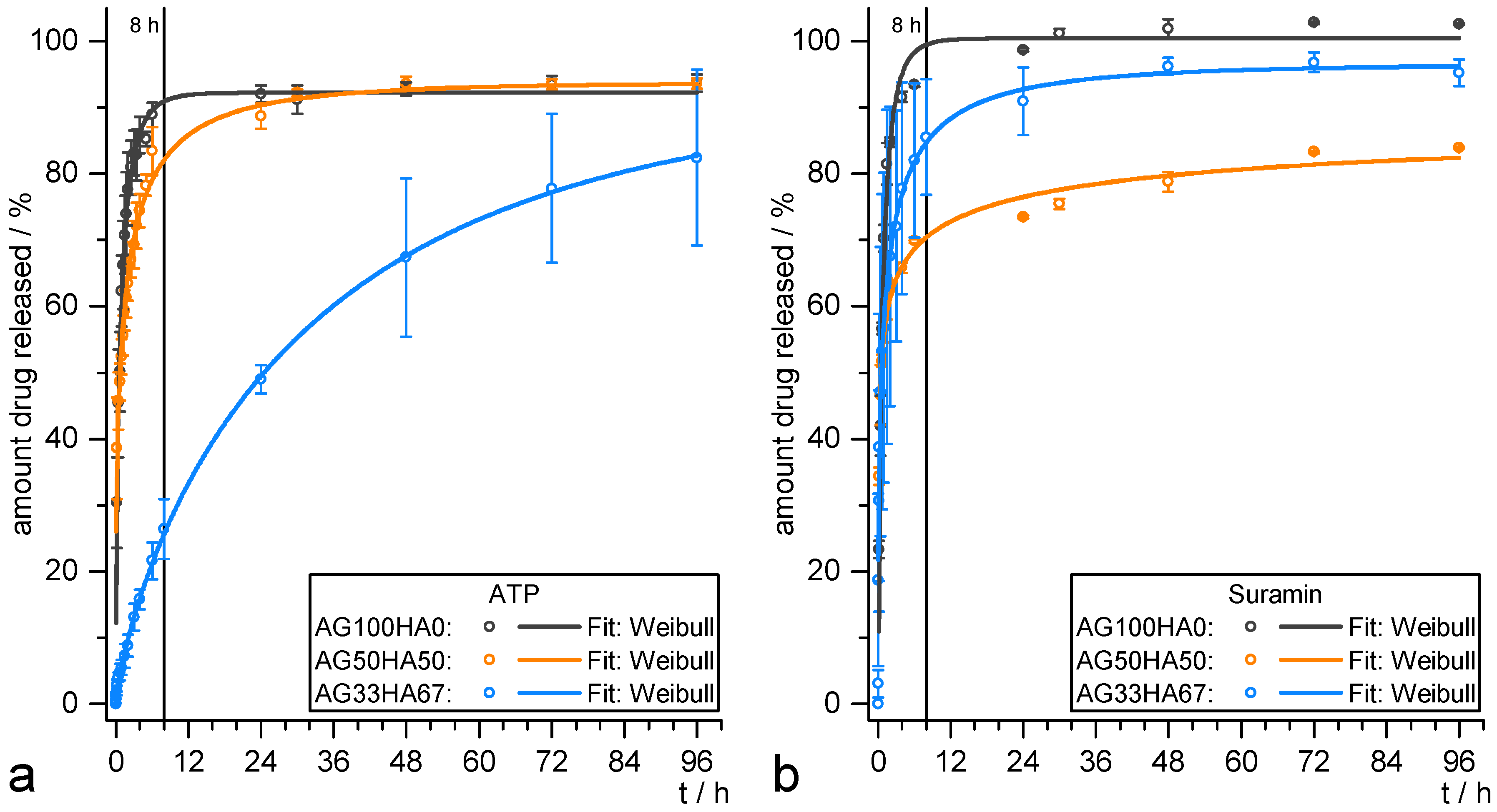
2.3. Release Studies
3. Materials and Methods
3.1. Preparation of Hydroxyapatite
3.2. Preparation of Agarose Gels
3.3. Preparation of AG/HA Composites
3.4. Characterization of Scaffolds
3.5. Biocompatibility Testing
3.6. Drug Loading and Drug Release Test
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine 5′-triphosphate |
| BET | Brunauer–Emmett–Teller surface analysis |
| EDX | electron dispersive X-ray spectroscopy |
| HA | hydroxyapatite |
| hMSC | human mesenchymal stem cells |
| LYO | lyophilization |
| PBS | phosphate-buffered saline |
| SCD | supercritical drying |
| SEM | scanning electron microscopy |
| VD | vacuum dried |
Appendix A
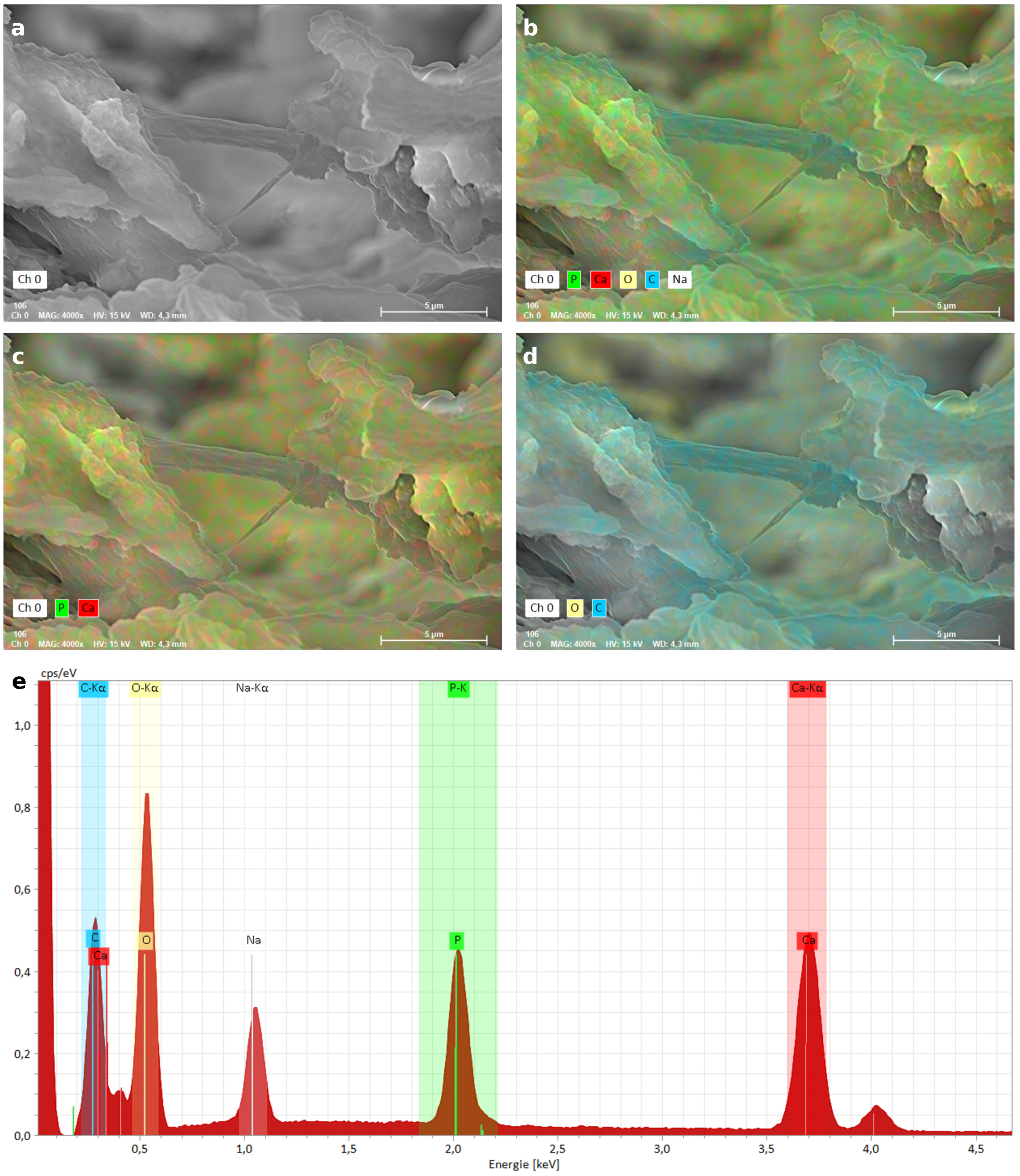
Appendix A.1. EDX Analysis

Appendix A.2. Swelling and Water Uptake Data

References
- Braddock, M.; Houston, P.; Campbell, C.; Ashcroft, P. Born Again Bone: Tissue Engineering for Bone Repair. Physiology 2001, 16, 208–213. [Google Scholar] [CrossRef]
- Spicer, P.P.; Kretlow, J.D.; Young, S.; Jansen, J.A.; Kasper, F.K.; Mikos, A.G. Evaluation of Bone Regeneration Using the Rat Critical Size Calvarial Defect. NIH Public Access 2012, 7, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Grotheer, V.; Schulze, M.; Tobiasch, E. Trends in Bone Tissue Engineering: Proteins for Osteogenic Differentiation and the Respective Scaffolding. In Purification—Principles and Trends; iConcept Press Ltd.: Hong Kong, China, 2014; pp. 1–43. [Google Scholar]
- Schulze, M.; Tobiasch, E. Artificial Scaffolds and Mesenchymal Stem Cells for Hard Tissues. In Tissue Engineering III: Cell-Surface Interactions for Tissue Culture; Kasper, C., Witte, F., Pörtner, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 153–194. [Google Scholar]
- Rezwan, K.; Chen, Q.; Blaker, J.; Boccaccini, A.R. Biodegradable and Bioactive Porous Polymer/Inorganic Composite Scaffolds for Bone Tissue Engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem Cells: Past, Present, and Future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, A.; Lukomska, B.; Janowski, M. Concise Review: Mesenchymal Stem Cells: From Roots to Boost. Stem Cells 2019, 37. [Google Scholar] [CrossRef]
- Zippel, N.; Limbach, C.A.; Ratajski, N.; Urban, C.; Luparello, C.; Pansky, A.; Kassack, M.U.; Tobiasch, E. Purinergic Receptors Influence the Differentiation of Human Mesenchymal Stem Cells. Stem Cells Dev. 2012, 21, 884–900. [Google Scholar] [CrossRef]
- Ciciarello, M.; Zini, R.; Rossi, L.; Salvestrini, V.; Ferrari, D.; Manfredini, R.; Lemoli, R.M. Extracellular Purines Promote the Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells to the Osteogenic and Adipogenic Lineages. Stem Cells Dev. 2013, 22, 1097–1111. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef]
- Ottensmeyer, P.; Witzler, M.; Schulze, M.; Tobiasch, E. Small Molecules Enhance Scaffold-Based Bone Grafts via Purinergic Receptor Signaling in Stem Cells. Int. J. Mol. Sci. 2018, 19, 3601. [Google Scholar] [CrossRef]
- Leiendecker, A.; Witzleben, S.; Schulze, M.; Tobiasch, E. Template-Mediated Biomineralization for Bone Tissue Engineering. Curr. Stem Cell Res. Ther. 2016, 12, 103–123. [Google Scholar] [CrossRef]
- Witzler, M.; Alzagameem, A.; Bergs, M.; Khaldi-Hansen, B.E.; Klein, S.E.; Hielscher, D.; Kamm, B.; Kreyenschmidt, J.; Tobiasch, E.; Schulze, M. Lignin-Derived Biomaterials for Drug Release and Tissue Engineering. Molecules 2018, 23, 1885. [Google Scholar] [CrossRef]
- Leach, J.K.; Whitehead, J. Materials-Directed Differentiation of Mesenchymal Stem Cells for Tissue Engineering and Regeneration. ACS Biomater. Sci. Eng. 2018, 4, 1115–1127. [Google Scholar] [CrossRef]
- Quelch, K.J.; Melick, R.; Bingham, P.J.; Mercuri, S.M. Chemical Composition of Human Bone. Arch. Oral Biol. 1983, 28, 665–674. [Google Scholar] [CrossRef]
- Palmer, L.C.; Newcomb, C.J.; Kaltz, S.R.; Spoerke, E.D.; Stupp, S.I. Biomimetic Systems for Hydroxyapatite Mineralization Inspired By Bone and Enamel. Chem. Rev. 2008, 108, 4754–4783. [Google Scholar] [CrossRef]
- Ginebra, M.P.; Canal, C.; Espanol, M.; Pastorino, D.; Montufar, E.B. Calcium Phosphate Cements as Drug Delivery Materials. Adv. Drug Deliv. Rev. 2012, 64, 1090–1110. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef]
- Götz, W.; Tobiasch, E.; Witzleben, S.; Schulze, M. Effects of Silicon Compounds on Biomineralization, Osteogenesis, and Hard Tissue Formation. Pharmaceutics 2019, 11, 117. [Google Scholar] [CrossRef]
- Babczyk, P.; Conzendorf, C.; Klose, J.; Schulze, M.; Harre, K.; Tobiasch, E. Stem Cells on Biomaterials for Synthetic Grafts to Promote Vascular Healing. J. Clin. Med. 2014, 3, 39–87. [Google Scholar] [CrossRef]
- Vega, S.; Kwon, M.; Burdick, J. Recent Advances in Hydrogels for Cartilage Tissue Engineering. Eur. Cells Mater. 2017, 33, 59–75. [Google Scholar] [CrossRef]
- Gkioni, K.; Leeuwenburgh, S.C.G.; Douglas, T.E.L.; Mikos, A.G.; Jansen, J.A. Mineralization of Hydrogels for Bone Regeneration. Tissue Eng. Part B Rev. 2010, 16, 577–585. [Google Scholar] [CrossRef]
- Arnott, S.; Fulmer, A.; Scott, W.; Dea, I.; Moorhouse, R.; Rees, D. The Agarose Double Helix and its Function in Agarose Gel Structure. J. Mol. Biol. 1974, 90, 269–284. [Google Scholar] [CrossRef]
- Normand, V.; Lootens, D.L.; Amici, E.; Plucknett, K.P. New Insight into Agarose Gel Mechanical Properties. Biomacromolecules 2000, 1, 730–738. [Google Scholar] [CrossRef]
- Zarrintaj, P.; Manouchehri, S.; Ahmadi, Z.; Saeb, M.R.; Urbanska, A.M.; Kaplan, D.L.; Mozafari, M. Agarose-based biomaterials for tissue engineering. Carbohydr. Polym. 2018, 187, 66–84. [Google Scholar] [CrossRef]
- Tabata, M.; Shimoda, T.; Sugihara, K.; Ogomi, D.; Serizawa, T.; Akashi, M. Osteoconductive and Hemostatic Properties of Apatite Formed on/in Agarose Gel as a Bone-Grafting Material. J. Biomed. Mater. Res. 2003, 67B, 680–688. [Google Scholar] [CrossRef]
- Schipper, D.; Babczyk, P.; Elsayed, F.; Klein, S.E.; Schulze, M.; Tobiasch, E. Chapter 21—The Effect of Nanostructured Surfaces on Stem Cell Fate. In Nanostructures for Novel Therapy; Ficai, D., Grumezescu, A.M., Eds.; Micro and Nano Technologies, Elsevier: Amsterdam, The Netherlands, 2017; pp. 567–589. [Google Scholar]
- Suzawa, Y.; Funaki, T.; Watanabe, J.; Iwai, S.; Yura, Y.; Nakano, T.; Umakoshi, Y.; Akashi, M. Regenerative Behavior of Biomineral/Agarose Composite Gels as Bone Grafting Materials in Rat Cranial Defects. J. Biomed. Mater. Res. Part A 2010, 93, 965–975. [Google Scholar] [CrossRef]
- Suzawa, Y.; Kubo, N.; Iwai, S.; Yura, Y.; Ohgushi, H.; Akashi, M. Biomineral/Agarose Composite Gels Enhance Proliferation of Mesenchymal Stem Cells with Osteogenic Capability. Int. J. Mol. Sci. 2015, 16, 14245–14258. [Google Scholar] [CrossRef]
- Kolanthai, E.; Dikeshwar Colon, V.S.; Sindu, P.A.; Chandra, V.S.; Karthikeyan, K.R.; Babu, M.S.; Sundaram, S.M.; Palanichamy, M.; Kalkura, S.N. Effect of Solvent; Enhancing the Wettability and Engineering the Porous Structure of a Calcium Phosphate/Agarose Composite for Drug Delivery. RSC Adv. 2015, 5, 18301–18311. [Google Scholar] [CrossRef]
- Khanarian, N.T.; Haney, N.M.; Burga, R.A.; Lu, H.H. A Functional Agarose-Hydroxyapatite Scaffold for Osteochondral Interface Regeneration. Biomaterials 2012, 33, 5247–5258. [Google Scholar] [CrossRef]
- Sánchez-Salcedo, S.; Nieto, A.; Vallet-Regí, M. Hydroxyapatite/β-tricalcium Phosphate/Agarose Macroporous Scaffolds for Bone Tissue Engineering. Chem. Eng. J. 2008, 137, 62–71. [Google Scholar] [CrossRef]
- Paris, J.; Román, J.; Manzano, M.; Cabañas, M.; Vallet-Regí, M. Tuning Dual-Drug Release From Composite Scaffolds for Bone Regeneration. Int. J. Pharm. 2015, 486, 30–37. [Google Scholar] [CrossRef]
- Iwai, S.; Shimizu, H.; Suzawa, Y.; Akashi, M.; Yura, Y. Hydroxyapatite Agarose Composite Gels as a Biochemical Material for the Repair of Alveolar Bone Defects Due to Cleft Lip and Palate. J. Oral Maxillofac. Surg. Med. Pathol. 2014, 27, 637–644. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, Y.; Tong, H.; Shen, X.; Chen, L.; Ran, J. A Detailed Study of Homogeneous Agarose/Hydroxyapatite Nanocomposites for Load-Bearing Bone Tissue. Int. J. Biol. Macromol. 2016, 82, 134–143. [Google Scholar] [CrossRef]
- Raina, D.B.; Larsson, D.; Mrkonjic, F.; Isaksson, H.; Kumar, A.; Lidgren, L.; Tägil, M. Gelatin- Hydroxyapatite- Calcium Sulphate Based Biomaterial for Long Term Sustained Delivery of Bone Morphogenic Protein-2 and Zoledronic Acid for Increased Bone Formation: In-Vitro and In-Vivo Carrier Properties. J. Control. Release 2018, 272, 83–96. [Google Scholar] [CrossRef]
- He, X.; Liu, Y.; Yuan, X.; Lu, L. Enhanced Healing of Rat Calvarial Defects With MSCs Loaded on BMP-2 Releasing Chitosan/Alginate/Hydroxyapatite Scaffolds. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Kolanthai, E.; Abinaya Sindu, P.; Thanigai Arul, K.; Sarath Chandra, V.; Manikandan, E.; Narayana Kalkura, S. Agarose Encapsulated Mesoporous Carbonated Hydroxyapatite Nanocomposites Powder for Drug Delivery. J. Photochem. Photobiol. B Biol. 2017, 166, 220–231. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kalkura, S.; Palanichamy, M.; Arivuoli, D.; Dierks, K.; Bocelli, G.; Betzel, C. Synthesis of Stoichiometric Nano Crystalline Hydroxyapatite by Ethanol-Based Sol–Gel Technique at Low Temperature. J. Cryst. Growth 2004, 263, 517–523. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Antonakos, A.; Liarokapis, E.; Leventouri, T. Micro-Raman and FTIR Studies of Synthetic and Natural Apatites. Biomaterials 2007, 28, 3043–3054. [Google Scholar] [CrossRef]
- Ren, F.; Ding, Y.; Leng, Y. Infrared Spectroscopic Characterization of Carbonated Apatite: A Combined Experimental and Computational Study. J. Biomed. Mater. Res. Part A 2014, 102, 496–505. [Google Scholar] [CrossRef]
- Román, J.; Cabañas, M.V.; Peña, J.; Vallet-Regí, M. Control of the Pore Architecture in Three-Dimensional Hydroxyapatite-Reinforced Hydrogel Scaffolds. Sci. Technol. Adv. Mater. 2011, 12, 045003. [Google Scholar] [CrossRef]
- Kolanthai, E.; Ganesan, K.; Epple, M.; Kalkura, S.N. Synthesis of Nanosized Hydroxyapatite/Agarose Powders for Bone Filler and Drug Delivery Application. Mater. Today Commun. 2016, 8, 31–40. [Google Scholar] [CrossRef]
- García-González, C.; Alnaief, M.; Smirnova, I. Polysaccharide-Based Aerogels—Promising Biodegradable Carriers for Drug Delivery Systems. Carbohydr. Polym. 2011, 86, 1425–1438. [Google Scholar] [CrossRef]
- Robitzer, M.; Tourrette, A.; Horga, R.; Valentin, R.; Boissière, M.; Devoisselle, J.; Di Renzo, F.; Quignard, F. Nitrogen Sorption as a Tool for the Characterisation of Polysaccharide Aerogels. Carbohydr. Polym. 2011, 85, 44–53. [Google Scholar] [CrossRef]
- Reverchon, E.; Pisanti, P.; Cardea, S. Nanostructured PLLA-Hydroxyapatite Scaffolds Produced by a Supercritical Assisted Technique. Ind. Eng. Chem. Res. 2009, 48, 5310–5316. [Google Scholar] [CrossRef]
- Velasco, M.A.; Narváez-Tovar, C.A.; Garzón-Alvarado, D.A. Design, Materials, and Mechanobiology of Biodegradable Scaffolds for Bone Tissue Engineering. BioMed Res. Int. 2015, 2015, 1–21. [Google Scholar] [CrossRef]
- Oyen, M.L. Mechanical Characterisation of Hydrogel Materials. Int. Mater. Rev. 2014, 59, 44–59. [Google Scholar] [CrossRef]
- DeKosky, B.J.; Dormer, N.H.; Ingavle, G.C.; Roatch, C.H.; Lomakin, J.; Detamore, M.S.; Gehrke, S.H. Hierarchically Designed Agarose and Poly(Ethylene Glycol) Interpenetrating Network Hydrogels for Cartilage Tissue Engineering. Tissue Eng. Part C Methods 2010, 16, 1533–1542. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D Biomaterial Scaffolds and Osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef]
- Mehdizadeh, H.; Sumo, S.; Bayrak, E.S.; Brey, E.M.; Cinar, A. Three-Dimensional Modeling of Angiogenesis in Porous Biomaterial Scaffolds. Biomaterials 2013, 34, 2875–2887. [Google Scholar] [CrossRef]
- Bruschi, M.L. Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015. [Google Scholar]
- Gelardi, G.; Mantellato, S.; Marchon, D.; Palacios, M.; Eberhardt, A.; Flatt, R. Chemistry of Chemical Admixtures. In Science and Technology of Concrete Admixtures; Elsevier: Amsterdam, The Netherlands, 2016; pp. 149–218. [Google Scholar]
- Landi, E.; Tampieri, A.; Celotti, G.; Sprio, S. Densification Behaviour and Mechanisms of Synthetic Hydroxyapatites. J. Eur. Ceram. Soc. 2000, 20, 2377–2387. [Google Scholar] [CrossRef]
- Román, J.; Cabañas, M.; Peña, J.; Doadrio, J.; Vallet-Regí, M. An Optimized β-tricalcium Phosphate and Agarose Scaffold Fabrication Technique. J. Biomed. Mater. Res. Part A 2008, 84, 99–107. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AG100HA0 | AG50HA50 | AG33HA67 | ||||
|---|---|---|---|---|---|---|
| LYO | SCD | LYO | SCD | LYO | SCD | |
| Drug | Material | Model | ||
|---|---|---|---|---|
| Weibull | First-Order | Korsmeyer–Peppas | ||
| ATP | AG100HA0 | |||
| h−1 | ||||
| AG50HA50 | ||||
| h−1 | ||||
| AG33HA67 | ||||
| h−1 | ||||
| suramin | AG100HA0 | |||
| h−1 | ||||
| AG50HA50 | ||||
| h−1 | ||||
| AG33HA67 | ||||
| h−1 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witzler, M.; Ottensmeyer, P.F.; Gericke, M.; Heinze, T.; Tobiasch, E.; Schulze, M. Non-Cytotoxic Agarose/Hydroxyapatite Composite Scaffolds for Drug Release. Int. J. Mol. Sci. 2019, 20, 3565. https://doi.org/10.3390/ijms20143565
Witzler M, Ottensmeyer PF, Gericke M, Heinze T, Tobiasch E, Schulze M. Non-Cytotoxic Agarose/Hydroxyapatite Composite Scaffolds for Drug Release. International Journal of Molecular Sciences. 2019; 20(14):3565. https://doi.org/10.3390/ijms20143565
Chicago/Turabian StyleWitzler, Markus, Patrick Frank Ottensmeyer, Martin Gericke, Thomas Heinze, Edda Tobiasch, and Margit Schulze. 2019. "Non-Cytotoxic Agarose/Hydroxyapatite Composite Scaffolds for Drug Release" International Journal of Molecular Sciences 20, no. 14: 3565. https://doi.org/10.3390/ijms20143565
APA StyleWitzler, M., Ottensmeyer, P. F., Gericke, M., Heinze, T., Tobiasch, E., & Schulze, M. (2019). Non-Cytotoxic Agarose/Hydroxyapatite Composite Scaffolds for Drug Release. International Journal of Molecular Sciences, 20(14), 3565. https://doi.org/10.3390/ijms20143565