HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension



Abstract
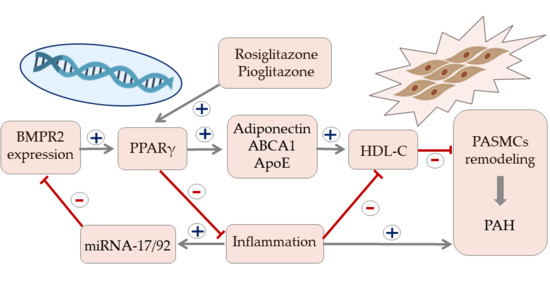
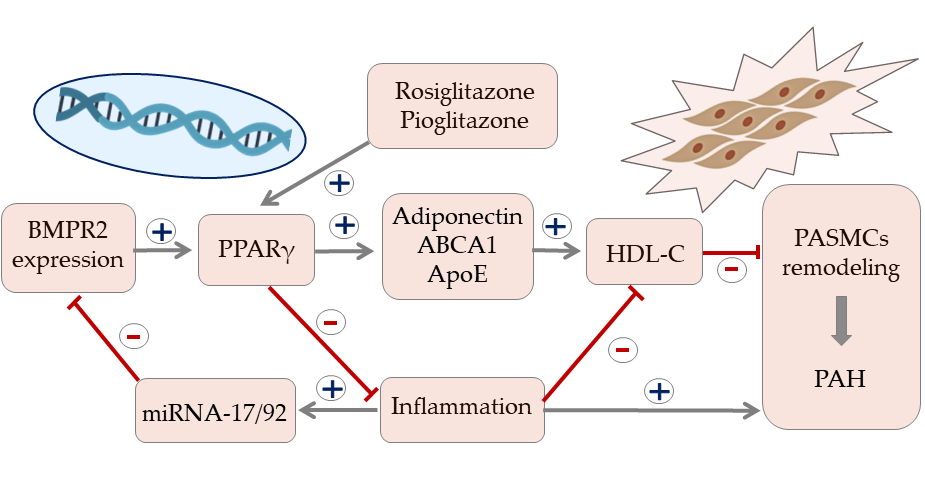
1. Protective Role of HDL in the Systemic Circulation
2. Pulmonary Arterial Hypertension
3. HDL-Mediated Mechanisms of Cardiovascular Protection in Pulmonary Hypertension
4. HDL in NO and Prostacyclin Pathway
5. HDL and Inflammation
6. HDL and Insulin Resistance
7. HDL and Platelet Activation
8. HDL and microRNA
9. HDL and Pulmonary Vascular Disease Severity
10. Summary
Funding
Conflicts of Interest
References
- Kannel, W.B.; Dawber, T.R.; Friedman, G.D.; Glennon, W.E.; McNamara, P.M. Risk factors in coronary heart disease. An evaluation of several serum lipids as predictors of coronary heart disease; the framingham study. Ann. Intern. Med. 1964, 61, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Badimon, J.J.; Badimon, L.; Fuster, V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J. Clin. Invest. 1990, 85, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.M.; Krauss, R.M.; Spangler, E.A.; Verstuyft, J.G.; Clift, S.M. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991, 353, 265–267. [Google Scholar] [CrossRef]
- Toth, P.P.; Barter, P.J.; Rosenson, R.S.; Boden, W.E.; Chapman, M.J.; Cuchel, M.; D’Agostino, R.B., Sr.; Davidson, M.H.; Davidson, W.S.; Heinecke, J.W.; et al. High-density lipoproteins: A consensus statement from the national lipid association. J. Clin. Lipidol. 2013, 7, 484–525. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C., Jr.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the american college of cardiology/american heart association task force on practice guidelines. Circulation 2014, 129, S49–S73. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; deGoma, E.M. Approach to the patient with extremely low HDL-cholesterol. J. Clin. Endocrinol. Metab. 2012, 97, 3399–3407. [Google Scholar] [CrossRef]
- Hovingh, G.K.; de Groot, E.; van der Steeg, W.; Boekholdt, S.M.; Hutten, B.A.; Kuivenhoven, J.A.; Kastelein, J.J. Inherited disorders of HDL metabolism and atherosclerosis. Curr. Opin. Lipidol. 2005, 16, 139–145. [Google Scholar] [CrossRef]
- Santos, R.D.; Asztalos, B.F.; Martinez, L.R.; Miname, M.H.; Polisecki, E.; Schaefer, E.J. Clinical presentation, laboratory values, and coronary heart disease risk in marked high-density lipoprotein-deficiency states. J. Clin. Lipidol. 2008, 2, 237–247. [Google Scholar] [CrossRef]
- Oldoni, F.; Sinke, R.J.; Kuivenhoven, J.A. Mendelian disorders of high-density lipoprotein metabolism. Circ. Res. 2014, 114, 124–142. [Google Scholar] [CrossRef]
- De Grooth, G.J.; Klerkx, A.H.; Stroes, E.S.; Stalenhoef, A.F.; Kastelein, J.J.; Kuivenhoven, J.A. A review of CETP and its relation to atherosclerosis. J. Lipid. Res. 2004, 45, 1967–1974. [Google Scholar] [CrossRef]
- Sirtori, C.R.; Calabresi, L.; Franceschini, G.; Baldassarre, D.; Amato, M.; Johansson, J.; Salvetti, M.; Monteduro, C.; Zulli, R.; Muiesan, M.L.; et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: The limone sul garda study. Circulation 2001, 103, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Canner, P.L.; Berge, K.G.; Wenger, N.K.; Stamler, J.; Friedman, L.; Prineas, R.J.; Friedewald, W. Fifteen year mortality in coronary drug project patients: Long-term benefit with niacin. J. Am. Coll. Cardiol. 1986, 8, 1245–1255. [Google Scholar] [CrossRef]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Group, H.T.C.; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Group, H.T.R.C.; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Wu, Z.; Lou, Y.; Qiu, X.; Liu, Y.; Lu, L.; Chen, Q.; Jin, W. Association of cholesteryl ester transfer protein (CETP) gene polymorphism, high density lipoprotein cholesterol and risk of coronary artery disease: A meta-analysis using a Mendelian randomization approach. BMC Med. Genet. 2014, 15, 118. [Google Scholar] [CrossRef]
- Ray, K.K.; Ditmarsch, M.; Kallend, D.; Niesor, E.J.; Suchankova, G.; Upmanyu, R.; Anzures-Cabrera, J.; Lehnert, V.; Pauly-Evers, M.; Holme, I.; et al. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: the dal-ACUTE randomized trial. Eur. Heart J. 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Meriwether, D.; Lee, Y.Y.; Shahbazian, A.; Reddy, S.T. High levels of oxidized fatty acids in HDL are associated with impaired HDL function in patients with active rheumatoid arthritis. Clin. Rheumatol. 2018, 37, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (ESC) and the european respiratory society (ERS)endorsed by: Association for european paediatric and congenital cardiology (AEPC), international society for heart and lung transplantation (ISHLT). Eur. Heart J. 2015, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 2012, 122, 4306–4313. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaïci, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Foreman, A.J.; Frost, A.E.; Badesch, D.B.; Benton, W.W.; McGoon, M.D. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: A registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) analysis. J. Heart Lung. Transplant. 2015, 34, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Pittrow, D.; Ghofrani, H.A.; Opitz, C.F.; Huscher, D.; Hoeper, M.M. International, prospective register for the documentation of first-line and maintenance therapy in patients with pulmonary hypertension (CompERA-XL). Dtsch. Med. Wochenschr 2009, 134 (Suppl. 5), S173–S175. [Google Scholar] [CrossRef]
- Kylhammar, D.; Kjellström, B.; Hjalmarsson, C.; Jansson, K.; Nisell, M.; Söderberg, S.; Rådegran, G. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur. Heart J. 2017, 39, 4175–4181. [Google Scholar] [CrossRef]
- Boucly, A.; Weatherald, J.; Humbert, M.; Sitbon, O. Risk assessment in pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, F.; Nigro, E.; Mollica, M.; Costigliola, A.; D’Agnano, V.; Daniele, A.; Bianco, A.; Guerra, G. Pulmonary hypertension and obesity: Focus on adiponectin. Int. J. Mol. Sci. 2019, 20, 912. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Meloche, J.; Paulin, R.; Boucherat, O.; Provencher, S.; Bonnet, S. DNA damage and pulmonary hypertension. Int. J. Mol. Sci. 2016, 17, 990. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Kopec, G.; Waligora, M.; Tyrka, A.; Jonas, K.; Pencina, M.J.; Zdrojewski, T.; Moertl, D.; Stokwiszewski, J.; Zagozdzon, P.; Podolec, P. Low-density lipoprotein cholesterol and survival in pulmonary arterial hypertension. Sci. Rep. 2017, 7, 41650. [Google Scholar] [CrossRef] [PubMed]
- Kopeć, G.; Moertl, D.; Jankowski, P.; Tyrka, A.; Sobień, B.; Podolec, P. Pulmonary artery pulse wave velocity in idiopathic pulmonary arterial hypertension. Can. J. Cardiol. 2013, 29, 683–690. [Google Scholar] [CrossRef]
- Jonas, K.; Waligóra, M.; Magoń, W.; Zdrojewski, T.; Stokwiszewski, J.; Płazak, W.; Podolec, P.; Kopeć, G. Prognostic role of traditional cardiovascular risk factors in patients with idiopathic pulmonary arterial hypertension. Arch. Med. Sci. 2018, 14. [Google Scholar] [CrossRef]
- Brunner, N.W.; Skhiri, M.; Fortenko, O.; Hsi, A.; Haddad, F.; Khazeni, N.; Zamanian, R.T. Impact of insulin resistance on ventricular function in pulmonary arterial hypertension. J. Heart Lung. Transplant. 2014, 33, 721–726. [Google Scholar] [CrossRef]
- Rohrer, L.; Ohnsorg, P.M.; Lehner, M.; Landolt, F.; Rinninger, F.; von Eckardstein, A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ. Res. 2009, 104, 1142–1150. [Google Scholar] [CrossRef]
- Kontush, A. HDL-mediated mechanisms of protection in cardiovascular disease. Cardiovasc. Res. 2014, 103, 341–349. [Google Scholar] [CrossRef]
- Nofer, J.R.; van der Giet, M.; Tolle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Godecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Invest. 2004, 113, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Nakajima, H.; Toh, R.; Hirata, K.I.; Ishida, T. Cardioprotective effects of high-density lipoprotein beyond its anti-atherogenic action. J. Atheroscler. Thromb. 2018, 25, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef]
- Li, X.A.; Titlow, W.B.; Jackson, B.A.; Giltiay, N.; Nikolova-Karakashian, M.; Uittenbogaard, A.; Smart, E.J. High density lipoprotein binding to scavenger receptor, Class B, type I activates endothelial nitric-oxide synthase in a ceramide-dependent manner. J. Biol. Chem. 2002, 277, 11058–11063. [Google Scholar] [CrossRef] [PubMed]
- Hardy, L.M.; Frisdal, E.; Le Goff, W. Critical role of the human ATP-binding cassette G1 transporter in cardiometabolic diseases. Int. J. Mol. Sci. 2017, 18, 1892. [Google Scholar] [CrossRef] [PubMed]
- Ferre, R.; Aragones, G.; Plana, N.; Merino, J.; Heras, M.; Buixadera, C.; Masana, L. High-density lipoprotein cholesterol and apolipoprotein A1 levels strongly influence the reactivity of small peripheral arteries. Atherosclerosis 2011, 216, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J. Lipid Res. 1999, 40, 345–353. [Google Scholar]
- Gruaz, L.; Delucinge-Vivier, C.; Descombes, P.; Dayer, J.M.; Burger, D. Blockade of T cell contact-activation of human monocytes by high-density lipoproteins reveals a new pattern of cytokine and inflammatory genes. PLoS ONE 2010, 5, e9418. [Google Scholar] [CrossRef]
- Inoue, M.; Niki, M.; Ozeki, Y.; Nagi, S.; Chadeka, E.A.; Yamaguchi, T.; Osada-Oka, M.; Ono, K.; Oda, T.; Mwende, F.; et al. High-density lipoprotein suppresses tumor necrosis factor alpha production by mycobacteria-infected human macrophages. Sci. Rep. 2018, 8, 6736. [Google Scholar] [CrossRef]
- De Nardo, D.; Labzin, L.I.; Kono, H.; Seki, R.; Schmidt, S.V.; Beyer, M.; Xu, D.; Zimmer, S.; Lahrmann, C.; Schildberg, F.A.; et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2013, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Grun, J.L.; Manjarrez-Reyna, A.N.; Gomez-Arauz, A.Y.; Leon-Cabrera, S.; Ruckert, F.; Fragoso, J.M.; Bueno-Hernandez, N.; Islas-Andrade, S.; Melendez-Mier, G.; Escobedo, G. High-density lipoprotein reduction differentially modulates to classical and nonclassical monocyte subpopulations in metabolic syndrome patients and in LPS-stimulated primary human monocytes in vitro. J. Immunol. Res. 2018, 2018, 2737040. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.Q.; Li, Y.L.; Zhang, Y.X.; Xiao, Y.; Li, Z.D.; Liu, L.Q.; Zhou, Y.S.; Zhang, H.Y.; Liu, Y.H.; Zhang, L.H.; et al. Sphingosine kinase 1/sphingosine 1-phosphate signalling pathway as a potential therapeutic target of pulmonary hypertension. Int. J. Clin. Exp. Med. 2015, 8, 11930–11935. [Google Scholar] [PubMed]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Obinata, H.; Conger, H.; Dahlback, B.; Kono, M.; Proia, R.L.; Smith, J.D.; et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal 2015, 8, ra79. [Google Scholar] [CrossRef] [PubMed]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.A.; Chapman, M.J.; Kontush, A. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ruffenach, G.; Umar, S.; Motayagheni, N.; Reddy, S.T.; Eghbali, M. Role of oxidized lipids in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.J.; Hough, G.; Hama, S.; Aboulhosn, J.; Belperio, J.A.; Saggar, R.; Van Lenten, B.J.; Ardehali, A.; Eghbali, M.; Reddy, S.; et al. Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm. Circ. 2015, 5, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Sugano, M.; Tsuchida, K.; Makino, N. High-density lipoproteins protect endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Biochem. Biophys. Res. Commun. 2000, 272, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.; Magon, W.; Podolec, P.; Kopec, G. Triglyceride-to-high-density lipoprotein cholesterol ratio and systemic inflammation in patients with idiopathic pulmonary arterial hypertension. Med. Sci. Monit. 2019, 25, 746–753. [Google Scholar] [CrossRef]
- Heresi, G.A.; Malin, S.K.; Barnes, J.W.; Tian, L.; Kirwan, J.P.; Dweik, R.A. Abnormal glucose metabolism and high-energy expenditure in idiopathic pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 2017, 14, 190–199. [Google Scholar]
- Barnes, J.W.; Tian, L.; Heresi, G.A.; Farver, C.F.; Asosingh, K.; Comhair, S.A.; Aulak, K.S.; Dweik, R.A. O-linked beta-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015, 131, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Stern, M.P.; Hazuda, H.P.; Mitchell, B.D.; Patterson, J.K. Cardiovascular risk factors in confirmed prediabetic individuals. Does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA 1990, 263, 2893–2898. [Google Scholar] [CrossRef]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Kopec, G.; Moertl, D.; Steiner, S.; Stepien, E.; Mikolajczyk, T.; Podolec, J.; Waligora, M.; Stepniewski, J.; Tomkiewicz-Pajak, L.; Guzik, T.; et al. Markers of thrombogenesis and fibrinolysis and their relation to inflammation and endothelial activation in patients with idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e82628. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczyk, R.; Kaminski, K. The role of platelets in the development and progression of pulmonary arterial hypertension. Adv. Med. Sci. 2018, 63, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Drew, B.G.; Ono, A.; Duffy, S.J.; Gordon, M.V.; Schoenwaelder, S.M.; Sviridov, D.; Cooper, M.E.; Kingwell, B.A.; Jackson, S.P. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation 2009, 120, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Nofer, J.R.; Brodde, M.F.; Kehrel, B.E. High-density lipoproteins, platelets and the pathogenesis of atherosclerosis. Clin. Exp. Pharmacol. Physiol. 2010, 37, 726–735. [Google Scholar] [CrossRef]
- Calabresi, L.; Gomaraschi, M.; Franceschini, G. Endothelial protection by high-density lipoproteins: From bench to bedside. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1724–1731. [Google Scholar] [CrossRef]
- Lee, A.; McLean, D.; Choi, J.; Kang, H.; Chang, W.; Kim, J. Therapeutic implications of microRNAs in pulmonary arterial hypertension. BMB Rep. 2014, 47, 311–317. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Girerd, B.; Montani, D.; Jais, X.; Eyries, M.; Yaici, A.; Sztrymf, B.; Savale, L.; Parent, F.; Coulet, F.; Godinas, L.; et al. Genetic counselling in a national referral centre for pulmonary hypertension. Eur. Respir. J. 2016, 47, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am. J. Respir. Crit. Care. Med. 2015, 192, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 2014, 3, 23743. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell. Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef]
- Meloche, J.; Le Guen, M.; Potus, F.; Vinck, J.; Ranchoux, B.; Johnson, I.; Antigny, F.; Tremblay, E.; Breuils-Bonnet, S.; Perros, F.; et al. miR-223 reverses experimental pulmonary arterial hypertension. Am. J. Physiol. Cell. Physiol. 2015, 309, C363–C372. [Google Scholar] [CrossRef]
- Smith, K.A.; Yuan, J.X.; Schumacker, P.T. MicroRNAs and PARP: co-conspirators with ROS in pulmonary hypertension. Focus on “miR-223 reverses experimental pulmonary arterial hypertension”. Am. J. Physiol. Cell. Physiol. 2015, 309, C361–C362. [Google Scholar] [CrossRef][Green Version]
- Sarrion, I.; Milian, L.; Juan, G.; Ramon, M.; Furest, I.; Carda, C.; Cortijo Gimeno, J.; Mata Roig, M. Role of circulating miRNAs as biomarkers in idiopathic pulmonary arterial hypertension: Possible relevance of miR-23a. Oxid. Med. Cell. Longev. 2015, 2015, 792846. [Google Scholar] [CrossRef]
- Heresi, G.A.; Aytekin, M.; Newman, J.; DiDonato, J.; Dweik, R.A. Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am. J. Respir. Crit. Care. Med. 2010, 182, 661–668. [Google Scholar] [CrossRef]
- Zhao, Q.H.; Peng, F.H.; Wei, H.; He, J.; Chen, F.D.; Di, R.M.; Jiang, X.; Jiang, R.; Chen, Y.J.; Heresi, G.A.; et al. Serum high-density lipoprotein cholesterol levels as a prognostic indicator in patients with idiopathic pulmonary arterial hypertension. Am. J. Cardiol. 2012, 110, 433–439. [Google Scholar] [CrossRef]
- Cracowski, J.L.; Labarere, J.; Renversez, J.C.; Degano, B.; Chabot, F.; Humbert, M. Plasma levels of high-density lipoprotein cholesterol are not associated with survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care. Med. 2012, 186, 107. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; McCully, R.B.; Murphy, J.G.; Kushwaha, S.S.; Frantz, R.P.; Kane, G.C. Usefulness of high-density lipoprotein cholesterol to predict survival in pulmonary arterial hypertension. Am. J. Cardiol. 2016, 118, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Aman, J.; Hovingh, G.K. Multilevel omics: A next step on the way to understanding pulmonary arterial hypertension? Thorax 2019, 74, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Harbaum, L.; Ghataorhe, P.; Wharton, J.; Jimenez, B.; Howard, L.S.G.; Gibbs, J.S.R.; Nicholson, J.K.; Rhodes, C.J.; Wilkins, M.R. Reduced plasma levels of small HDL particles transporting fibrinolytic proteins in pulmonary arterial hypertension. Thorax 2019, 74, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.; Magon, W.; Waligora, M.; Seweryn, M.; Podolec, P.; Kopec, G. High-density lipoprotein cholesterol level and pulmonary artery vasoreactivity in patients with idiopathic pulmonary arterial hypertension. Pol. Arch. Intern. Med. 2018, 128, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Khirfan, G.; Tejwani, V.; Wang, X.; Li, M.; DiDonato, J.; Dweik, R.A.; Smedira, N.; Heresi, G.A. Plasma levels of high density lipoprotein cholesterol and outcomes in chronic thromboembolic pulmonary hypertension. PLoS ONE 2018, 13, e0197700. [Google Scholar] [CrossRef] [PubMed]
- Camont, L.; Chapman, M.J.; Kontush, A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends. Mol. Med. 2011, 17, 594–603. [Google Scholar] [CrossRef]
- Rye, K.A.; Barter, P.J. Cardioprotective functions of HDLs. J. Lipid Res. 2014, 55, 168–179. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Type of Pulmonary Hypertension | Pathogenesis | Hemodynamic Characteristics | Specific Drug Therapy |
|---|---|---|---|
Pulmonary arterial hypertension
| Usually multifactorial including alterations in:
| PAPm ≥ 25 mmHg and PAWP ≤ 15 mmHg and PVR > 3 WU | Calcium channel blockers Endothelin receptor antagonists Phosphodiesterase type 5 inhibitors Guanylate cyclase stimulators Prostacyclin analogues Prostacyclin receptor agonists |
| Pulmonary hypertension due to left heart disease | Passive backward transmission of filling pressures from the left heart | PAPm ≥ 25 mmHg and PAWP > 15 mmHg | Global management of the underlying condition of the left heart |
| Pulmonary hypertension due to lung diseases and/or hypoxia | Alveolar hypoventilation, vascular remodeling, parenchymal destruction, and fibrosis | PAPm ≥ 25 mmHg | Treatment of the underlying lung disease, long-term oxygen therapy in hypoxemic patients |
| Chronic thromboembolic pulmonary hypertension | Obstructive pulmonary artery remodeling as a consequence of vessel thromboembolism | PAPm ≥ 25 mmHg and PAWP ≤ 15 mmHg | Pulmonary endarterectomy, balloon pulmonary angioplasty, targeted medical therapy |
| Pulmonary hypertension with unclear and/or multifactorial mechanisms | Mechanisms are multifactorial and usually poorly understood | PAPm ≥ 25 mmHg | Treatment is tailored for underlying diagnosis; treatment of pulmonary hypertension is secondary |
| Biological Activity of HDL | Potential Mechanisms of Vascular Protection |
|---|---|
| Vasodilatory activity |
|
| Anti-inflammatory properties |
|
| Antioxidative properties |
|
| Cytoprotection |
|
| Modulation of glucose metabolism |
|
| Regulation of platelet activation |
|
| Regulation of gene expression |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonas, K.; Kopeć, G. HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 20, 3514. https://doi.org/10.3390/ijms20143514
Jonas K, Kopeć G. HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2019; 20(14):3514. https://doi.org/10.3390/ijms20143514
Chicago/Turabian StyleJonas, Kamil, and Grzegorz Kopeć. 2019. "HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 20, no. 14: 3514. https://doi.org/10.3390/ijms20143514
APA StyleJonas, K., & Kopeć, G. (2019). HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 20(14), 3514. https://doi.org/10.3390/ijms20143514