Male-Specific Long Noncoding RNA TTTY15 Inhibits Non-Small Cell Lung Cancer Proliferation and Metastasis via TBX4

Abstract
1. Introduction
2. Results
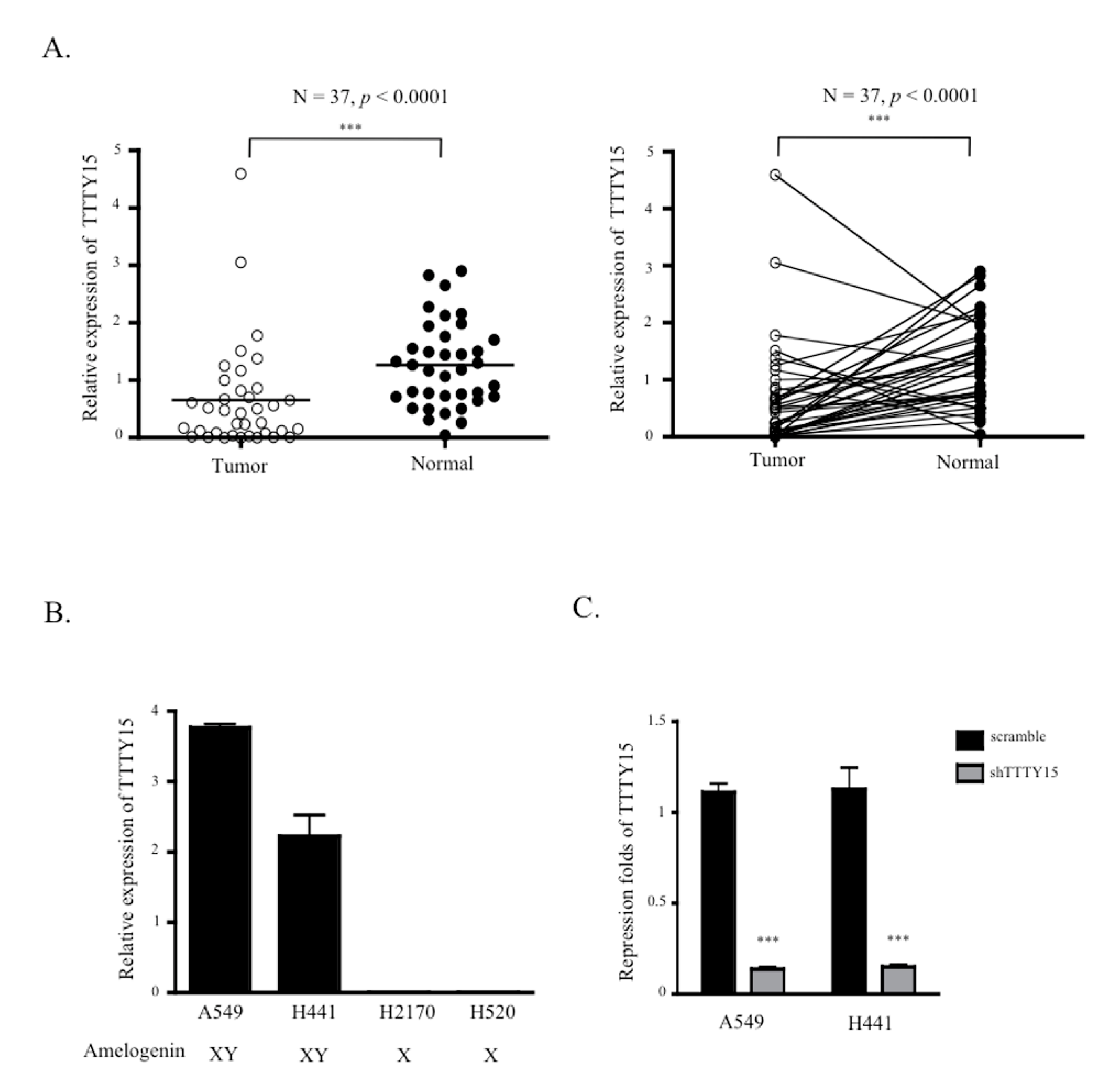
2.1. TTTY15 Expression is Low in NSCLC
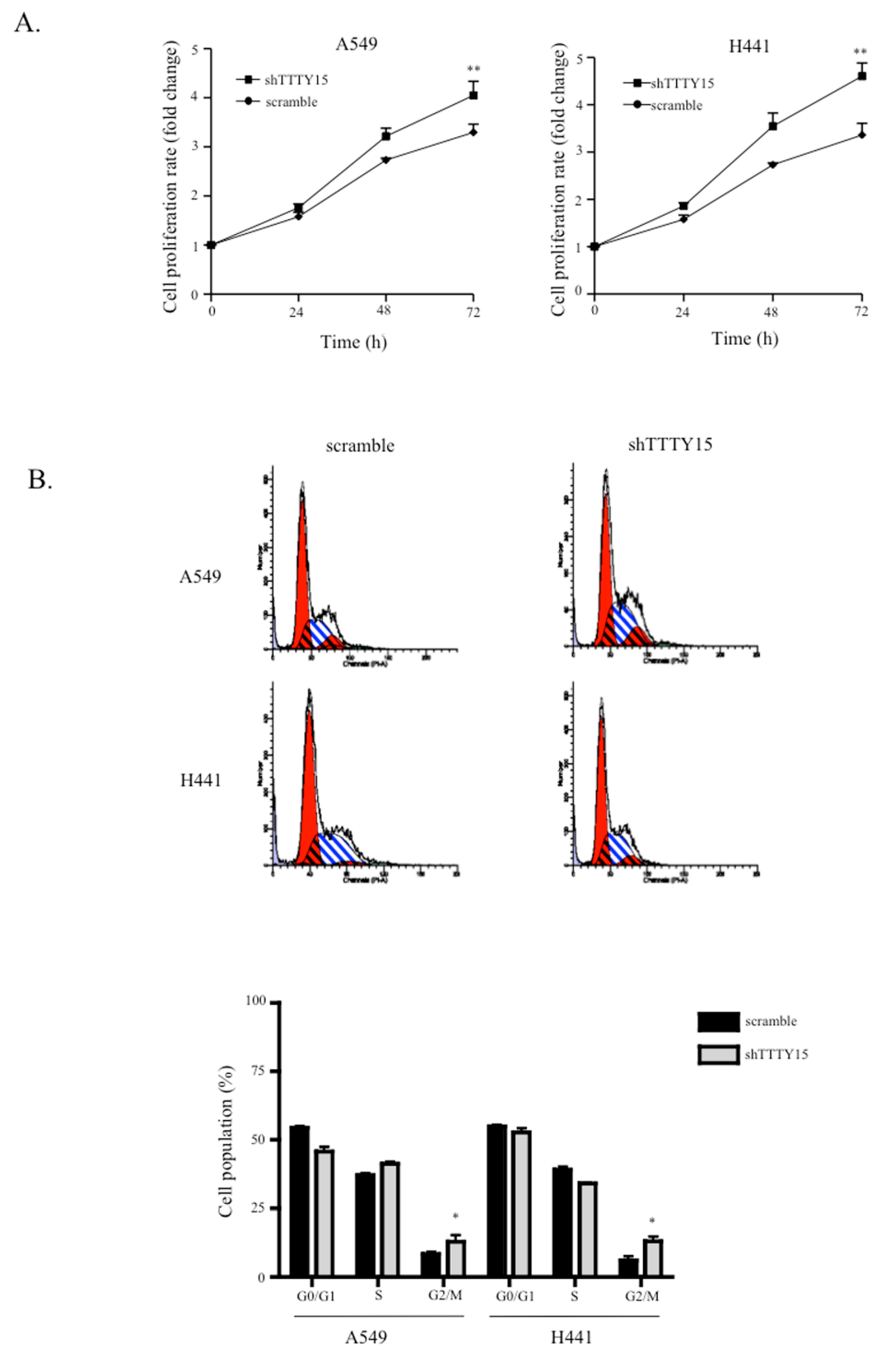
2.2. A Knockdown of TTTY15 Increased Lung Cancer Cells Proliferation and Cell Cycle Progression
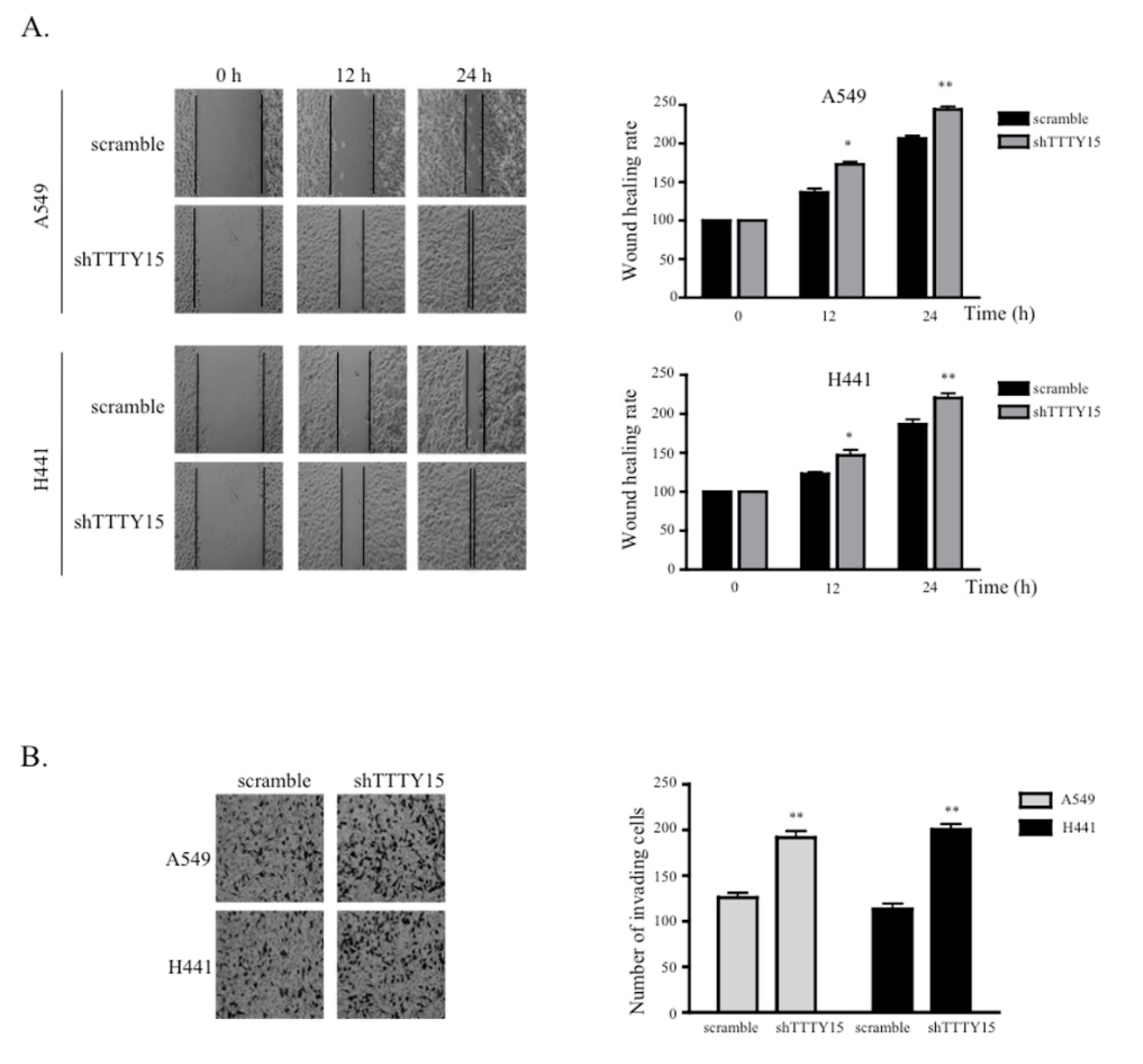
2.3. The Knockdown of TTTY15 Increased NSCLC Cell Migration and Invasion
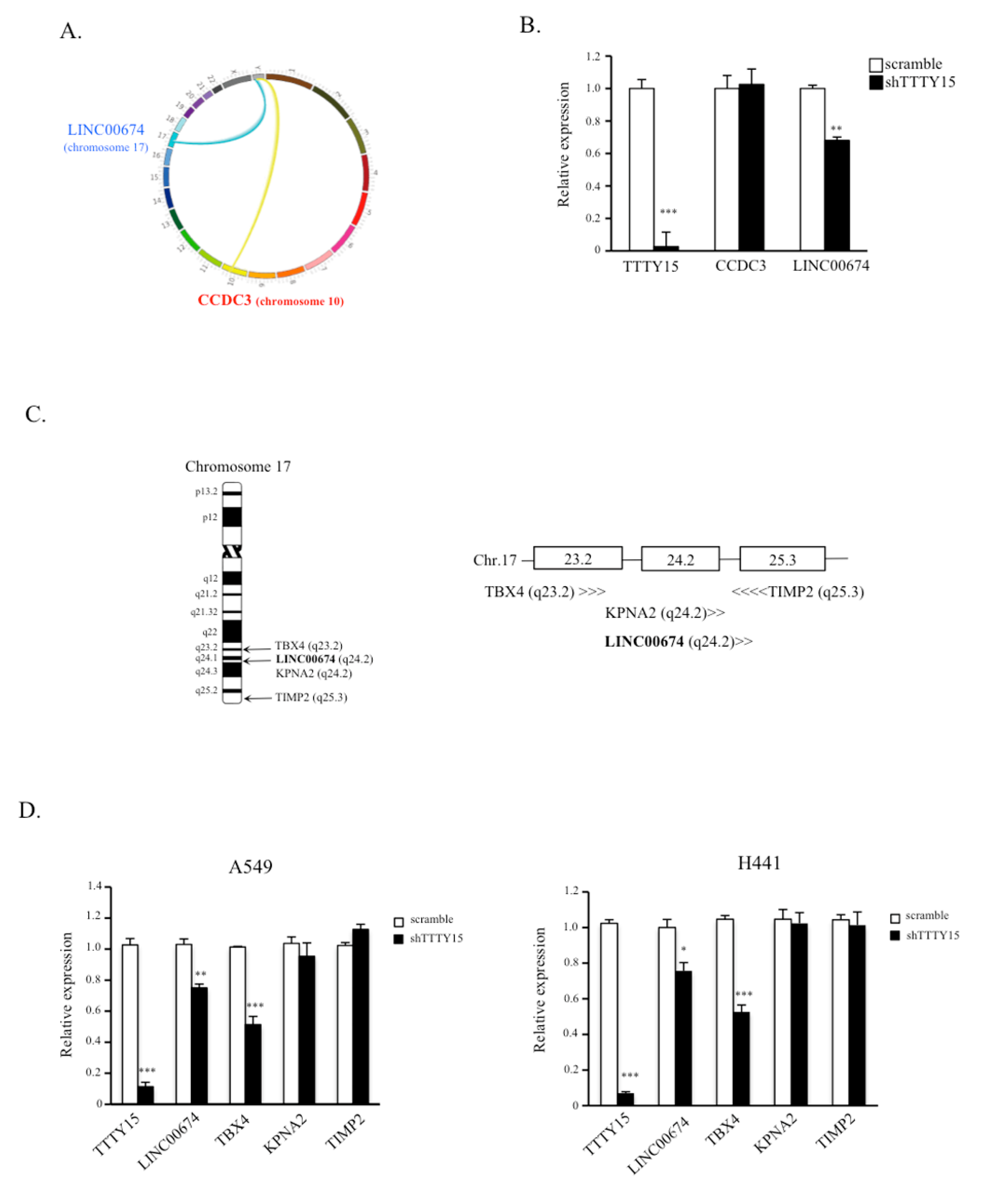
2.4. TBX4 Is a Potential Target Gene of TTTY15 and Is Positively Regulated by TTTY15
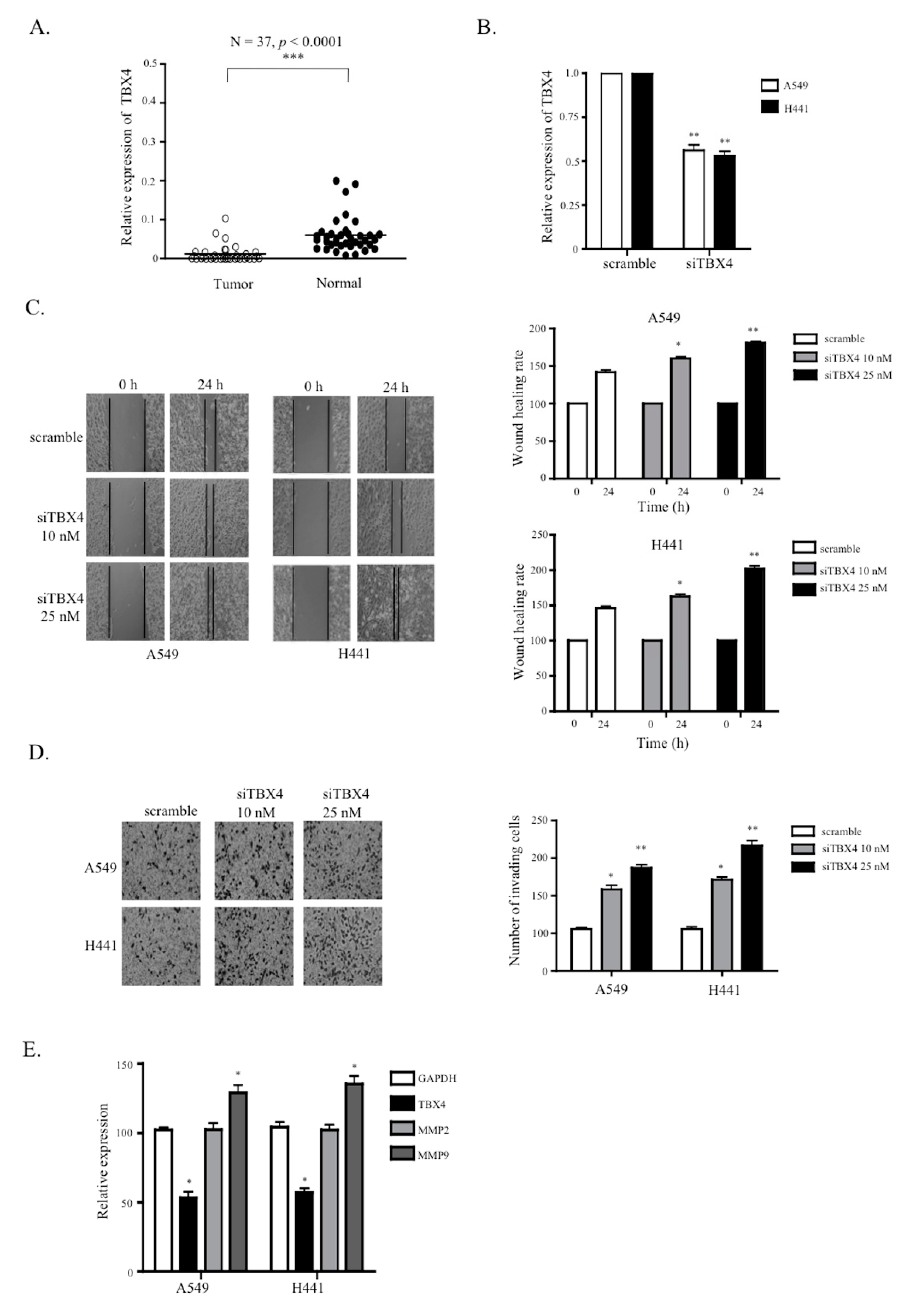
2.5. A Knockdown of TTTY15-Targeted TBX4 Increased Lung Cancer Cell Migration and Invasion
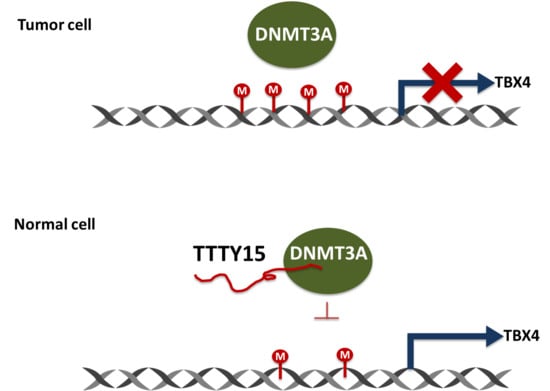
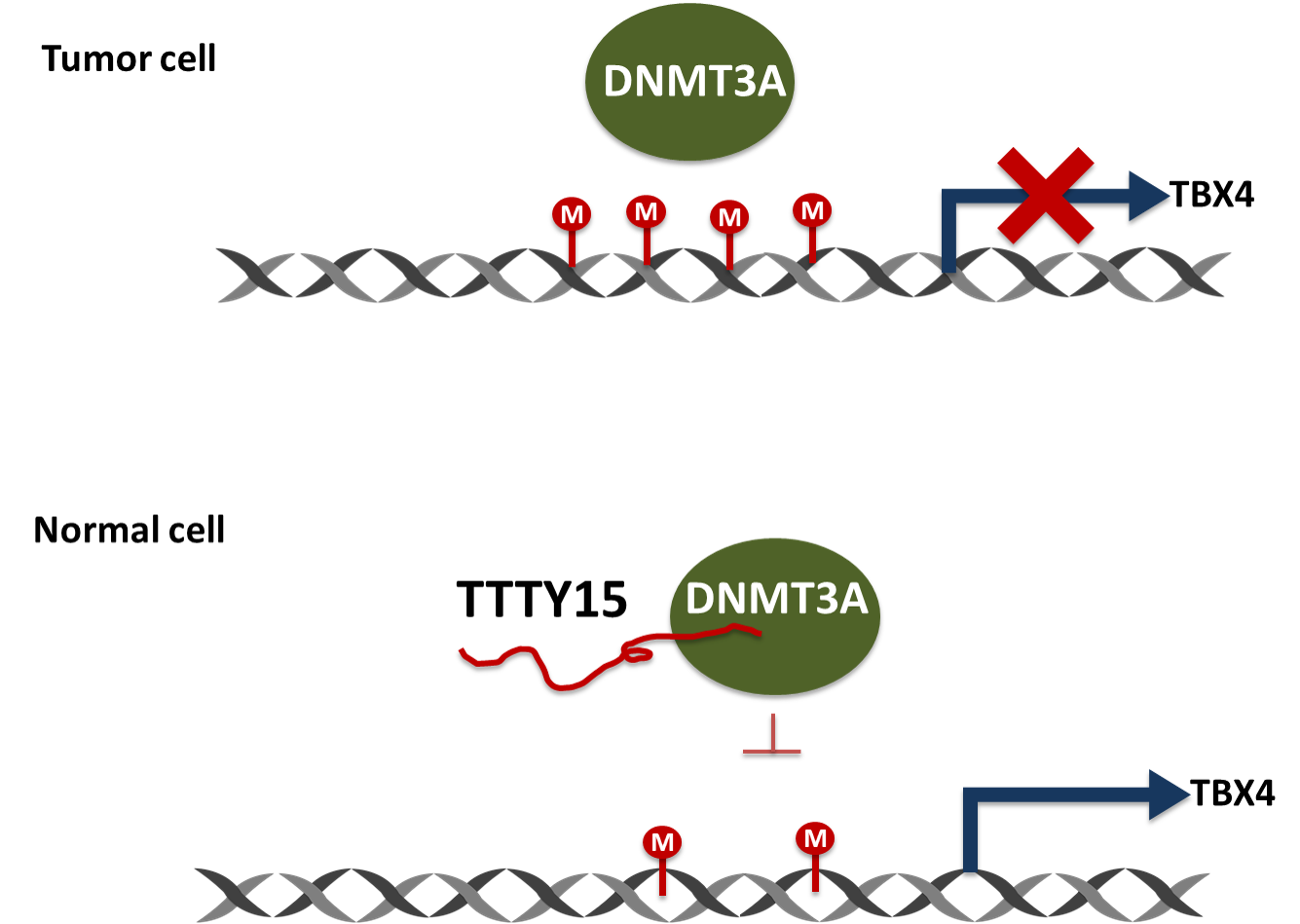
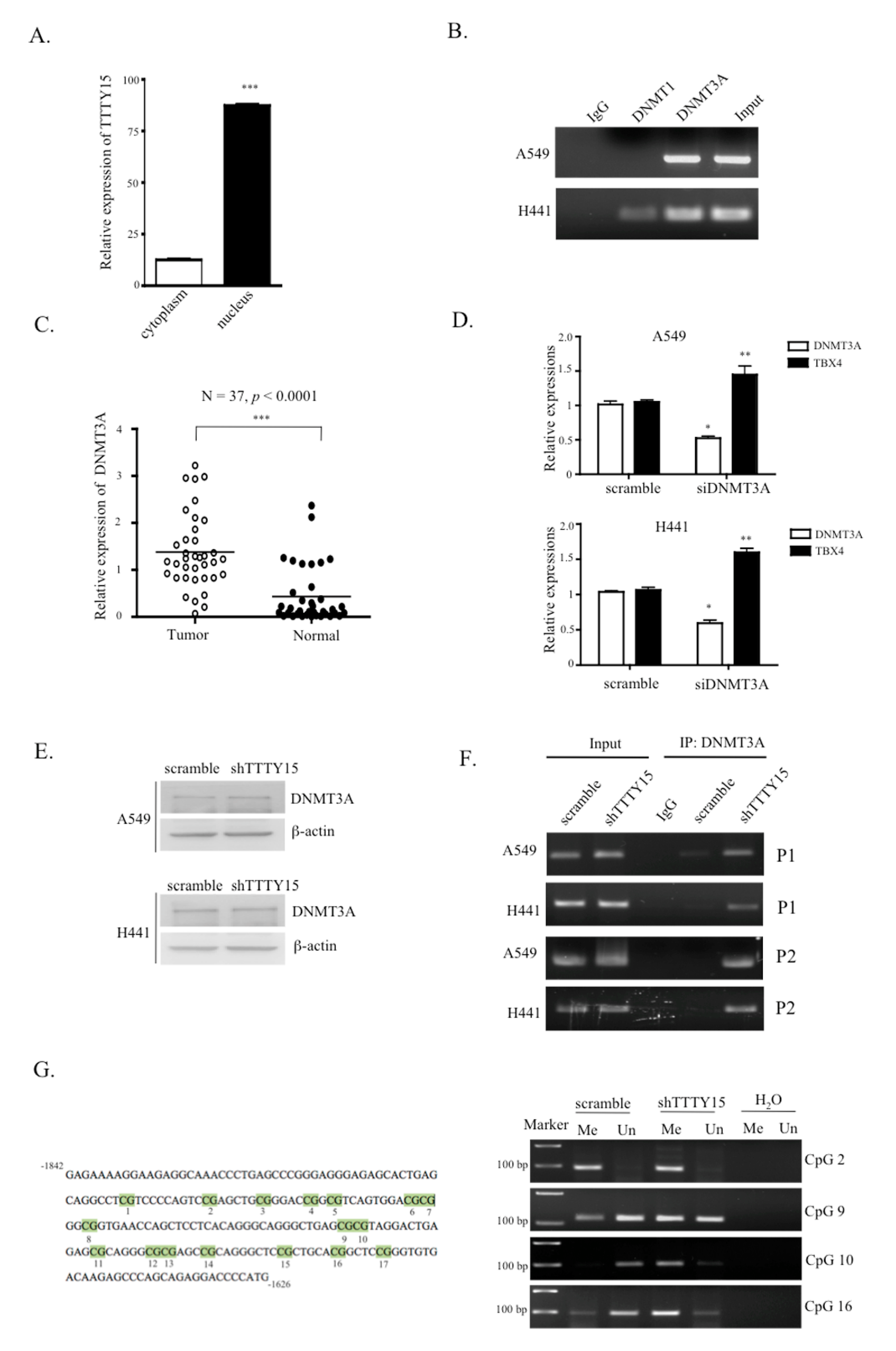
2.6. TTTY15 Is Associated with DNMT3A and Decreases Its Binding to the TBX4 Promoter
3. Discussion
4. Materials and Methods
4.1. Patients and Sample Collection
4.2. Cell Culture and Stable Transfection
4.3. SiRNA Transfection
4.4. Subcellular Fractionation, Total RNA Extraction, and qRT-PCR Analysis
4.5. A Cell Proliferation Assay
4.6. Wound Healing Scratch and Transwell Invasion Assays
4.7. Flow Cytometry for Cell Cycle Analysis
4.8. Circular Chromosome Conformation Capture
4.9. Next-Generation Sequencing and Bioinformatic Analysis of TTTY15 4C-Sequencing Data
4.10. The Western Blot Assay
4.11. RNA Immunoprecipitation
4.12. Chromatin Immunoprecipitation
4.13. Methylation-Specific PCR
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lundin, A.; Driscoll, B. Lung cancer stem cells: Progress and prospects. Cancer letters 2013, 338, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Dahlman, J.E.; Tammela, T.; Khan, O.F.; Sood, S.; Dave, A.; Cai, W.; Chirino, L.M.; Yang, G.R.; Bronson, R.; et al. Small RNA combination therapy for lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E3553–E3561. [Google Scholar] [CrossRef] [PubMed]
- Bareschino, M.A.; Schettino, C.; Rossi, A.; Maione, P.; Sacco, P.C.; Zeppa, R.; Gridelli, C. Treatment of advanced non small cell lung cancer. J. Thorac. Dis. 2011, 3, 122–133. [Google Scholar] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Sandberg, A.A. Chromosomal abnormalities and related events in prostate cancer. Hum. Pathol. 1992, 23, 368–380. [Google Scholar] [CrossRef]
- Lundgren, R.; Mandahl, N.; Heim, S.; Limon, J.; Henrikson, H.; Mitelman, F. Cytogenetic analysis of 57 primary prostatic adenocarcinomas. Genes Chromosomes Cancer 1992, 4, 16–24. [Google Scholar] [CrossRef]
- Sauter, G.; Moch, H.; Wagner, U.; Novotna, H.; Gasser, T.C.; Mattarelli, G.; Mihatsch, M.J.; Waldman, F.M. Y chromosome loss detected by FISH in bladder cancer. Cancer Genet. Cytogenet. 1995, 82, 163–169. [Google Scholar] [CrossRef]
- Wada, M.; Yokota, J.; Mizoguchi, H.; Terada, M.; Sugimura, T. Y chromosome abnormality in human stomach and lung cancer. Jpn. J. Cancer Res. 1987, 78, 780–783. [Google Scholar]
- Center, R.; Lukeis, R.; Vrazas, V.; Garson, O.M. Y chromosome loss and rearrangement in non-small-cell lung cancer. Int. J. Cancer 1993, 55, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Gramlich, T.; Abbott, K.; Varma, V. Y chromosome loss in esophageal carcinoma: An in situ hybridization study. Genes Chromosomes Cancer 1993, 8, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Dorak, M.T.; Karpuzoglu, E. Gender differences in cancer susceptibility: An inadequately addressed issue. Front. Genet. 2012, 3, 268. [Google Scholar] [CrossRef] [PubMed]
- Skaletsky, H.; Kuroda-Kawaguchi, T.; Minx, P.J.; Cordum, H.S.; Hillier, L.; Brown, L.G.; Repping, S.; Pyntikova, T.; Ali, J.; Bieri, T.; et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003, 423, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.; Koina, E.; Sankovic, N. How the gene content of human sex chromosomes evolved. Curr. Opin. Genet. Dev. 2006, 16, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Molina, E.; Chew, G.S.; Myers, S.A.; Clarence, E.M.; Eales, J.M.; Tomaszewski, M.; Charchar, F.J. A Novel Y-Specific Long Non-Coding RNA Associated with Cellular Lipid Accumulation in HepG2 cells and Atherosclerosis-related Genes. Sci. Rep. 2017, 7, 16710. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ren, S.; Jing, T.; Cai, X.; Liu, Y.; Wang, F.; Zhang, W.; Shi, X.; Chen, R.; Shen, J.; et al. Clinical utility of a novel urine-based gene fusion TTTY15-USP9Y in predicting prostate biopsy outcome. Urol. Oncol. 2015, 33, 384. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Peng, Z.; Mao, J.H.; Yu, Y.; Yin, C.; Gao, X.; Cui, Z.; Zhang, J.; Yi, K.; Xu, W.; et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012, 22, 806–821. [Google Scholar] [CrossRef] [PubMed]
- Zong, M.; Meng, M.; Li, L. Low expression of TBX4 predicts poor prognosis in patients with stage II pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2011, 12, 4953–4963. [Google Scholar] [CrossRef]
- Postma, D.S. Gender differences in asthma development and progression. Gender Med. 2007, 4, S133–S146. [Google Scholar] [CrossRef]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Luczak, E.D.; Leinwand, L.A. Sex-based cardiac physiology. Annu. Rev. Physiol. 2009, 71, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Ando, K.; Shinmyo, T.; Morita, K.; Mochizuki, A.; Kurimoto, N.; Tatsunami, S. Female gender is an independent prognostic factor in non-small-cell lung cancer: A meta-analysis. Ann. Thor. Cardiovasc. Surg. 2011, 17, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Fajkovic, H.; Halpern, J.A.; Cha, E.K.; Bahadori, A.; Chromecki, T.F.; Karakiewicz, P.I.; Breinl, E.; Merseburger, A.S.; Shariat, S.F. Impact of gender on bladder cancer incidence, staging, and prognosis. World J. Urol. 2011, 29, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhang, Y.; Mao, X.; Liu, X.; Mercer, E.; Marzec, J.; Ding, D.; Jiao, Y.; Qiu, Q.; Sun, Y.; et al. Transcription-mediated chimeric RNAs in prostate cancer: Time to revisit old hypothesis? Omics J. Integr. Biol. 2014, 18, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, Y.; Sun, Y.; Meng, L.; Xu, X. First case of AML with rare chromosome translocations: A case report of twins. BMC Cancer 2018, 18, 458. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tao, W.; Guo, Z.; Cao, J.; Huang, X. Suppression of long noncoding RNA TTTY15 attenuates hypoxia-induced cardiomyocytes injury by targeting miR-455-5p. Gene 2019, 701, 1–8. [Google Scholar] [CrossRef]
- Xiao, G.; Yao, J.; Kong, D.; Ye, C.; Chen, R.; Li, L.; Zeng, T.; Wang, L.; Zhang, W.; Shi, X.; et al. The Long Noncoding RNA TTTY15, Which Is Located on the Y Chromosome, Promotes Prostate Cancer Progression by Sponging let-7. Eur. Urol. 2018. [Google Scholar] [CrossRef]
- Spilianakis, C.G.; Lalioti, M.D.; Town, T.; Lee, G.R.; Flavell, R.A. Interchromosomal associations between alternatively expressed loci. Nature 2005, 435, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Lomvardas, S.; Barnea, G.; Pisapia, D.J.; Mendelsohn, M.; Kirkland, J.; Axel, R. Interchromosomal interactions and olfactory receptor choice. Cell 2006, 126, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Tsai, C.L.; Lee, J.T. Transient homologous chromosome pairing marks the onset of X inactivation. Science 2006, 311, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Bacher, C.P.; Guggiari, M.; Brors, B.; Augui, S.; Clerc, P.; Avner, P.; Eils, R.; Heard, E. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat. Cell Biol. 2006, 8, 293–299. [Google Scholar] [CrossRef]
- Galande, S.; Purbey, P.K.; Notani, D.; Kumar, P.P. The third dimension of gene regulation: Organization of dynamic chromatin loopscape by SATB1. Curr. Opin. Genet. Dev. 2007, 17, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Gondor, A.; Ohlsson, R. Chromosome crosstalk in three dimensions. Nature 2009, 461, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Misteli, T. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013, 20, 290–299. [Google Scholar] [CrossRef]
- Agulnik, S.I.; Garvey, N.; Hancock, S.; Ruvinsky, I.; Chapman, D.L.; Agulnik, I.; Bollag, R.; Papaioannou, V.; Silver, L.M. Evolution of mouse T-box genes by tandem duplication and cluster dispersion. Genetics 1996, 144, 249–254. [Google Scholar]
- Naiche, L.A.; Papaioannou, V.E. Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development 2003, 130, 2681–2693. [Google Scholar] [CrossRef]
- Arora, R.; Metzger, R.J.; Papaioannou, V.E. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet. 2012, 8, e1002866. [Google Scholar] [CrossRef]
- Xie, T.; Liang, J.; Liu, N.; Huan, C.; Zhang, Y.; Liu, W.; Kumar, M.; Xiao, R.; D’Armiento, J.; Metzger, D.; et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J. Clin. Investig. 2016, 126, 3626. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.I.; Litzinger, M.; Trono, P.; Hamilton, D.H.; Schlom, J.; Palena, C. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J. Clin. Investig. 2010, 120, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.; Bell, D.; Weber, R.S.; El-Naggar, A.K. CpG island methylation profiling in human salivary gland adenoid cystic carcinoma. Cancer 2011, 117, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J. A closer look at long-range chromosomal interactions. Trends Biochem. Sci. 2003, 28, 277–280. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 2005, 33, 259–271. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Bao, X.; Zhu, X.; Kwok, Y.K.; Sun, K.; Chen, X.; Huang, Y.; Jauch, R.; Esteban, M.A.; et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015, 25, 335–350. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, Q.; Cheng, J.; Qiu, X.; Zhang, J.; Fan, H. Depletion of DNMT3A suppressed cell proliferation and restored PTEN in hepatocellular carcinoma cell. J. Biomed. Biotechnol. 2010, 2010, 737535. [Google Scholar] [CrossRef]
- Deng, T.; Kuang, Y.; Wang, L.; Li, J.; Wang, Z.; Fei, J. An essential role for DNA methyltransferase 3a in melanoma tumorigenesis. Biochem. Biophys. Res. Commun. 2009, 387, 611–616. [Google Scholar] [CrossRef]
- Stadhouders, R.; Kolovos, P.; Brouwer, R.; Zuin, J.; van den Heuvel, A.; Kockx, C.; Palstra, R.J.; Wendt, K.S.; Grosveld, F.; van Ijcken, W.; et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat. Protocols 2013, 8, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.L., Jr.; Starmer, J.; Mugford, J.W.; Calabrese, J.M.; Mieczkowski, P.; Yee, D.; Magnuson, T. fourSig: A method for determining chromosomal interactions in 4C-Seq data. Nucleic Acids Res. 2014, 42, e68. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | n | Relative TTTY15 Expression | p Value | |
|---|---|---|---|---|
| Low (n = 27) | High (n = 10) | |||
| Age (years) | 0.714 | |||
| ≤65 | 18 | 14 | 4 | |
| >65 | 19 | 13 | 6 | |
| Tumor size (maximum diameter) | 0.453 | |||
| ≤3 cm | 14 | 9 | 5 | |
| >3 cm | 23 | 18 | 5 | |
| Lymph node metastasis | ||||
| N1 | 25 | 21 | 4 | 0.0486 |
| N0 | 12 | 6 | 6 | |
| TNM (Tumor-Node-Metastasis) stage | ||||
| I–II | 13 | 6 | 7 | 0.0167 |
| III–IV | 24 | 21 | 3 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, I.-L.; Chang, Y.-S.; Chan, W.-L.; Lee, Y.-T.; Yen, J.-C.; Yang, C.-A.; Hung, S.-Y.; Chang, J.-G. Male-Specific Long Noncoding RNA TTTY15 Inhibits Non-Small Cell Lung Cancer Proliferation and Metastasis via TBX4. Int. J. Mol. Sci. 2019, 20, 3473. https://doi.org/10.3390/ijms20143473
Lai I-L, Chang Y-S, Chan W-L, Lee Y-T, Yen J-C, Yang C-A, Hung S-Y, Chang J-G. Male-Specific Long Noncoding RNA TTTY15 Inhibits Non-Small Cell Lung Cancer Proliferation and Metastasis via TBX4. International Journal of Molecular Sciences. 2019; 20(14):3473. https://doi.org/10.3390/ijms20143473
Chicago/Turabian StyleLai, I-Lu, Ya-Sian Chang, Wen-Ling Chan, Ya-Ting Lee, Ju-Chen Yen, Chin-An Yang, Shih-Ya Hung, and Jan-Gowth Chang. 2019. "Male-Specific Long Noncoding RNA TTTY15 Inhibits Non-Small Cell Lung Cancer Proliferation and Metastasis via TBX4" International Journal of Molecular Sciences 20, no. 14: 3473. https://doi.org/10.3390/ijms20143473
APA StyleLai, I.-L., Chang, Y.-S., Chan, W.-L., Lee, Y.-T., Yen, J.-C., Yang, C.-A., Hung, S.-Y., & Chang, J.-G. (2019). Male-Specific Long Noncoding RNA TTTY15 Inhibits Non-Small Cell Lung Cancer Proliferation and Metastasis via TBX4. International Journal of Molecular Sciences, 20(14), 3473. https://doi.org/10.3390/ijms20143473