A Density Functional Theory (DFT) Study of the Acyl Migration Occurring during Lipase-Catalyzed Transesterifications


Abstract

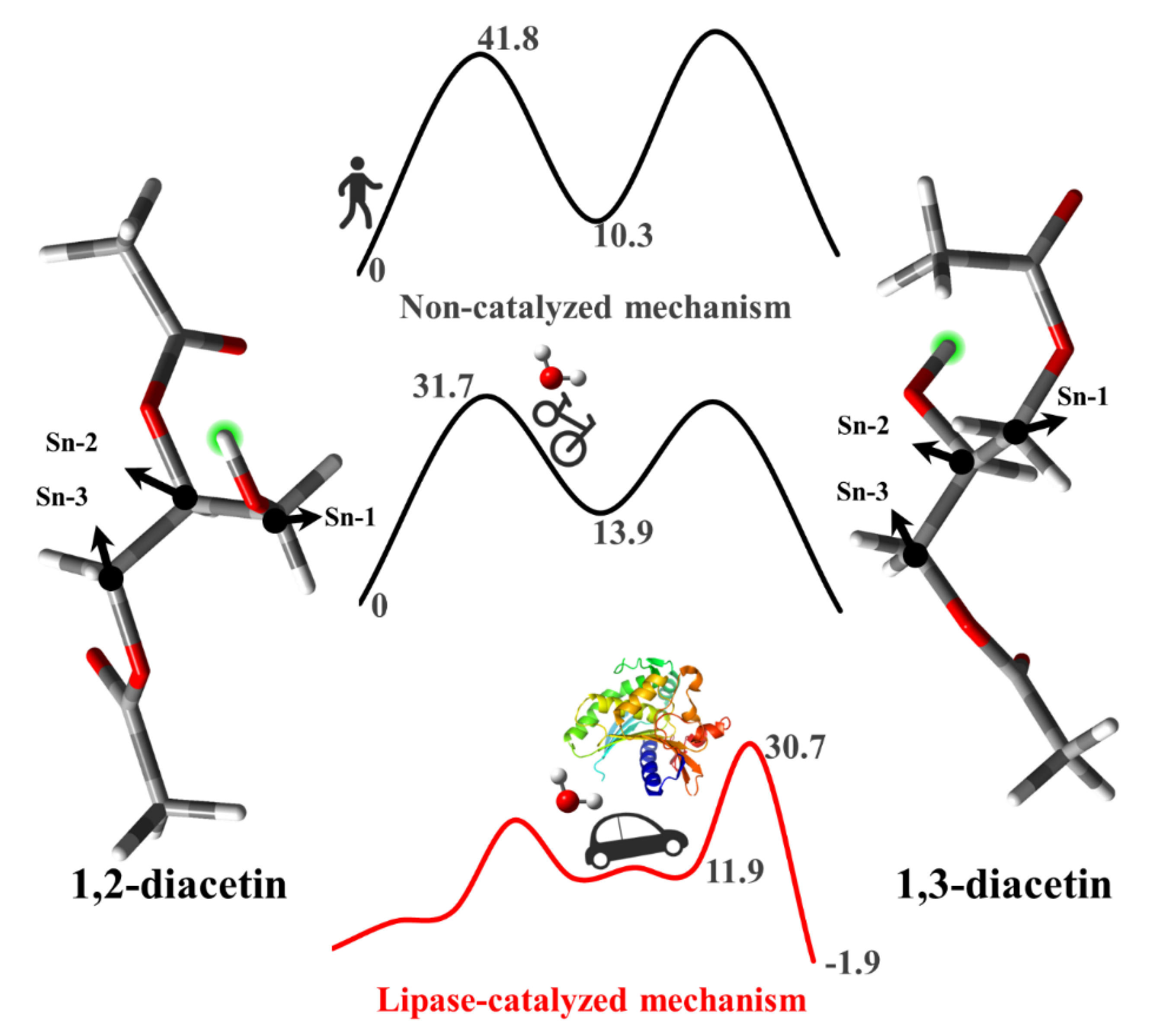{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Models and Details
2.1. Quantum Chemical Models
2.2. Computational Details
3. Results and Discussion
3.1. NCM of AM
3.2. LCM of AM
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, T.; Liu, L.; Hou, S.; Xu, J.; Yang, B.; Wang, Y.; Liu, J. Crystal structure of a mono—And diacylglycerol lipase from Malassezia globosa reveals a novel lid conformation and insights into the substrate specificity. J. Struct. Biol. 2012, 178, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147–148, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.; Crapiste, G.H.; Carrín, M.E. Study of acyl migration during enzymatic interesterification of liquid and fully hydrogenated soybean oil. J. Mol. Catal. B Enzym. 2015, 122, 117–124. [Google Scholar] [CrossRef]
- Kumar, A.; Dhar, K.; Kanwar, S.S.; Arora, P.K. Lipase catalysis in organic solvents: Advantages and applications. Biol. Proced. Online 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Kodali, D.R.; Tercyak, A.; Fahey, D.A.; Small, D.M. Acyl Migration in 1,2-Dipalmitoyl-Sn-Glycerol. Chem. Phys. Lipids 1990, 52, 163–170. [Google Scholar] [CrossRef]
- Liu, S.L.; Dong, X.Y.; Wei, F.; Wang, X.; Lv, X.; Wu, L.; Quek, S.Y.; Chen, H. Lipase Catalyzed Synthesis of ABA-Type Structured Lipid from Single Cell Oil and Tripalmitin. J. Food Process. Preserv. 2017, 41, e12843. [Google Scholar] [CrossRef]
- Yang, T.; Fruekilde, M.; Xu, X.B. Suppression of acyl migration in enzymatic production of structured lipids through temperature programming. Food Chem. 2005, 92, 101–107. [Google Scholar] [CrossRef]
- Mu, H.L.; Kurvinen, J.P.; Kallio, H.; Xu, X.B.; Høy, C.E. Quantitation of acyl migration during lipase-catalyzed acidolysis, and of the regioisomers of structured triacylglycerols formed. J. Am. Oil Chem. Soc. 2001, 78, 959–964. [Google Scholar] [CrossRef]
- Berk, Z. Physical Properties of Food Materials. Food Process Eng. Technol. 2013, 8, 1–27. [Google Scholar]
- Goh, S.; Yeong, S.; Wang, C. Transesterification of cocoa butter by fungal lipases: Effect of solvent on 1, 3-specificity. J. Am. Oil Chem. Soc. 1993, 70, 567. [Google Scholar] [CrossRef]
- Xu, X.B. Enzymatic production of structured lipids: Process reactions and acyl migration. Inform 2000, 11, 1121–1131. [Google Scholar]
- Xu, X.B.; Skands, A.R.H.; Hoy, C.E.; Mu, H.; Balchen, S.; Adler Nissen, J. Production of specific-structured lipids by enzymatic interesterification: Elucidation of acyl migration by response surface design. J. Am. Oil Chem. Soc. 1998, 75, 1179–1186. [Google Scholar] [CrossRef]
- Li, W.; Du, W.; Li, Q.; Li, R.W.; Liu, D.H. Dependence on the properties of organic solvent: Study on acyl migration kinetics of partial glycerides. Bioresour. Technol. 2010, 101, 5737–5742. [Google Scholar] [CrossRef] [PubMed]
- Svensson, J.; Adlercreutz, P. Effect of acyl migration in Lipozyme TL IM-catalyzed interesterification using a triacylglycerol model system. Eur. J. Lipid Sci. Technol. 2011, 113, 1258–1265. [Google Scholar] [CrossRef]
- Du, W.; Xu, Y.Y.; Liu, D.H.; Li, Z.B. Study on acyl migration in immobilized lipozyme TL-catalyzed transesterification of soybean oil for biodiesel production. J. Mol. Catal. B Enzym. 2005, 37, 68–71. [Google Scholar] [CrossRef]
- Melgosa, R.; Sanz, M.T.; Solaesa, Á.G.; De Paz, E.; Beltrán, S.; Lamas, D.L. Supercritical carbon dioxide as solvent in the lipase-catalyzed ethanolysis of fish oil: Kinetic study. J. CO2 Util. 2017, 17, 170–179. [Google Scholar] [CrossRef]
- Serdarevich, B. Glyceride isomerizations in lipid chemistry. J. Am. Oil Chem. Soc. 1967, 44, 381–393. [Google Scholar] [CrossRef]
- Lortie, R.; Trani, M.; Ergan, F. Kinetic study of the lipase-catalyzed synthesis of triolein. Biotechnol. Bioeng. 1993, 41, 1021–1026. [Google Scholar] [CrossRef]
- Josepha, L.; Davidl, C.; Karle, V. Acyl Migration Kinetics of Vegetable Oil 1,2-Diacylglycerols. J. Am. Oil Chem. Soc. 2008, 85, 307–312. [Google Scholar]
- Ferreira, M.L.; Tonetto, G.M. Enzymatic Synthesis of Structured Triglycerides: From Laboratory to Industry; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Himo, F. Quantum chemical modeling of enzyme active sites and reaction mechanisms. Theor. Chem. Acc. 2005, 116, 232–240. [Google Scholar] [CrossRef]
- Leščić Ašle, I.; Štefanić, Z.; Maršavelski, A.; Vianello, R.; Kojić-Prodić, B. Catalytic dyad in the SGNH hydrolase superfamily: In-depth insight into structural parameters tuning the catalytic process of extracellular lipase from Streptomyces rimosus. ACS Chem. Biol. 2017, 12, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Himo, F. Insights from Quantum Chemical Calculations into Active Site Structure and Reaction Mechanism of Manganese-Dependent Dinitrogenase Reductase-Activating Glycohydrolase; Stockholm University: Stockholm, Sweden, 2017. [Google Scholar]
- Brzozowski, A.M.; Derewenda, Z.S.; Dodson, E.J.; Dodson, G.G.; Turkenburg, J.P. Structure and Molecular-Model Refinement of Rhizomucor-Miehei Triacylglyceride Lipase—A Case-Study of the Use of Simulated Annealing in Partial Model Refinement. Acta Crystallogr. B 1992, 48, 307–319. [Google Scholar] [CrossRef]
- Norin, M.; Haeffner, F.; Achour, A.; Norin, T.; Hult, K. Computer Modeling of Substrate-Binding to Lipases from Rhizomucor-Miehei, Humicola-Lanuginosa, and Candida-Rugosa. Protein Sci. 1994, 3, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Mucsi, Z.; Szabó, A.; Hermecz, I.; Kucsman, Á.; Csizmadia, I.G. Modeling rate-controlling solvent effects. The pericyclic meisenheimer rearrangement of N-propargylmorpholine N-oxide. J. Am. Chem. Soc. 2005, 127, 7615–7631. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Mennucci, V.B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 (Revision A02); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Li, W.; Du, W.; Li, Q.; Sun, T.; Liu, D. Study on acyl migration kinetics of partial glycerides: Dependence on temperature and water activity. J. Mol. Catal. B Enzym. 2010, 63, 17–22. [Google Scholar] [CrossRef]
- Kroeger, A.A.; Karton, A. A Computational Investigation of the Uncatalysed and Water-Catalysed Acyl Rearrangements in Ingenol Esters. Aust. J. Chem. 2017, 71, 212–221. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Kreevoy, M.M.; Cleland, W.; Frey, P.A. Understanding enzymic catalysis: The importance of short, strong hydrogen bonds. Chem. Biol. 1997, 4, 259–267. [Google Scholar] [CrossRef]
- Guthrie, J.P. Short strong hydrogen bonds: Can they explain enzymic catalysis? Chem. Biol. 1996, 3, 163–170. [Google Scholar] [CrossRef]
- Schiøtt, B.; Iversen, B.B.; Hellerup Madsen, G.K.; Bruice, T.C. Characterization of the short strong hydrogen bond in benzoylacetone by ab initio calculations and accurate diffraction experiments. Implications for the electronic nature of low-barrier hydrogen bonds in enzymatic reactions. J. Am. Chem. Soc. 1998, 120, 12117–12124. [Google Scholar] [CrossRef]
- Ion, B.F.; Kazim, E.; Gauld, J.W. A Multi-Scale Computational Study on the Mechanism of Streptococcus pneumoniae Nicotinamidase (SpNic). Molecules 2014, 19, 15735–15753. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, J.; Hu, Z.; Hu, J.; Zhu, X.; Xiong, H. A Density Functional Theory (DFT) Study of the Acyl Migration Occurring during Lipase-Catalyzed Transesterifications. Int. J. Mol. Sci. 2019, 20, 3438. https://doi.org/10.3390/ijms20143438
Mao J, Hu Z, Hu J, Zhu X, Xiong H. A Density Functional Theory (DFT) Study of the Acyl Migration Occurring during Lipase-Catalyzed Transesterifications. International Journal of Molecular Sciences. 2019; 20(14):3438. https://doi.org/10.3390/ijms20143438
Chicago/Turabian StyleMao, Jinyuan, Zhenying Hu, Jiangning Hu, Xuemei Zhu, and Hua Xiong. 2019. "A Density Functional Theory (DFT) Study of the Acyl Migration Occurring during Lipase-Catalyzed Transesterifications" International Journal of Molecular Sciences 20, no. 14: 3438. https://doi.org/10.3390/ijms20143438
APA StyleMao, J., Hu, Z., Hu, J., Zhu, X., & Xiong, H. (2019). A Density Functional Theory (DFT) Study of the Acyl Migration Occurring during Lipase-Catalyzed Transesterifications. International Journal of Molecular Sciences, 20(14), 3438. https://doi.org/10.3390/ijms20143438