Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
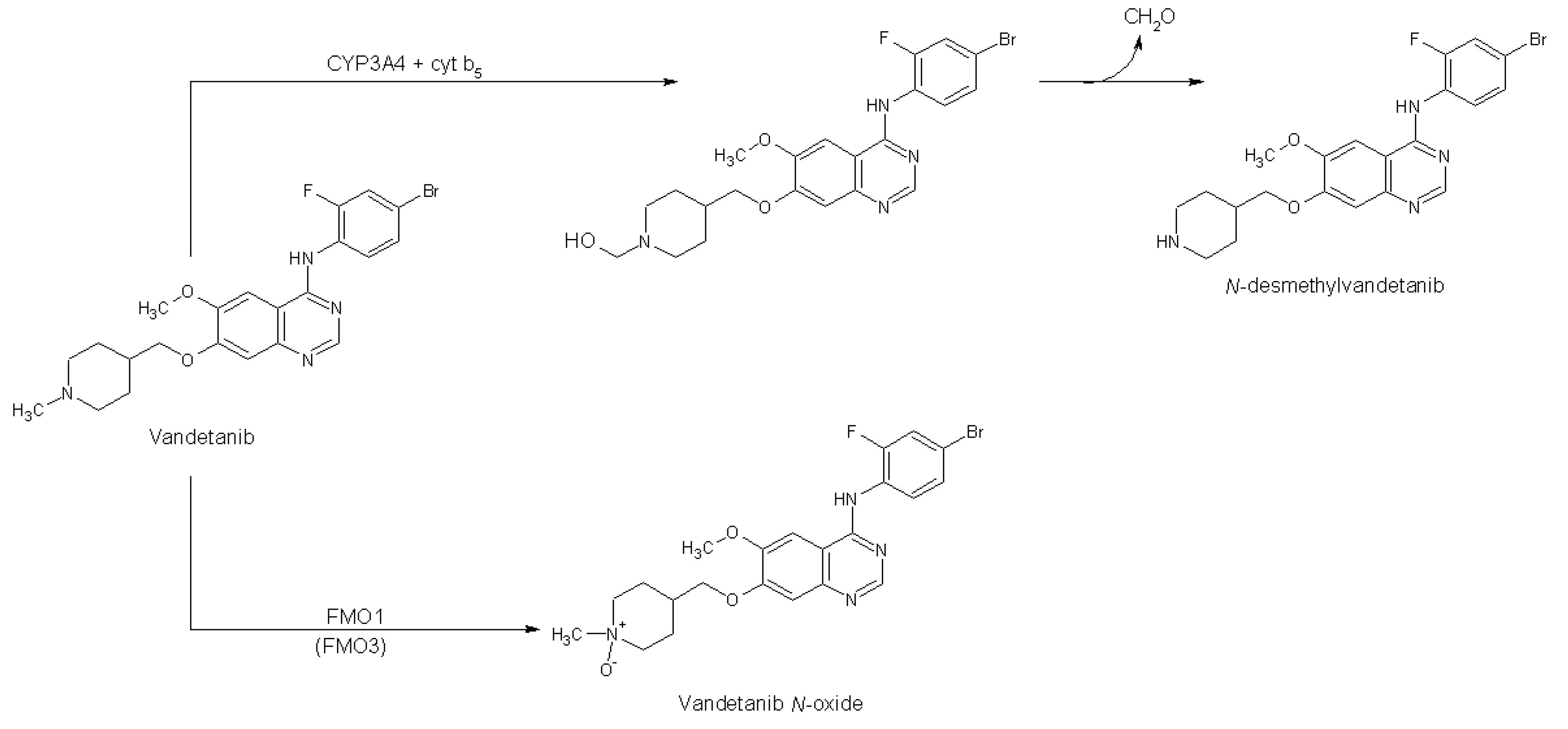
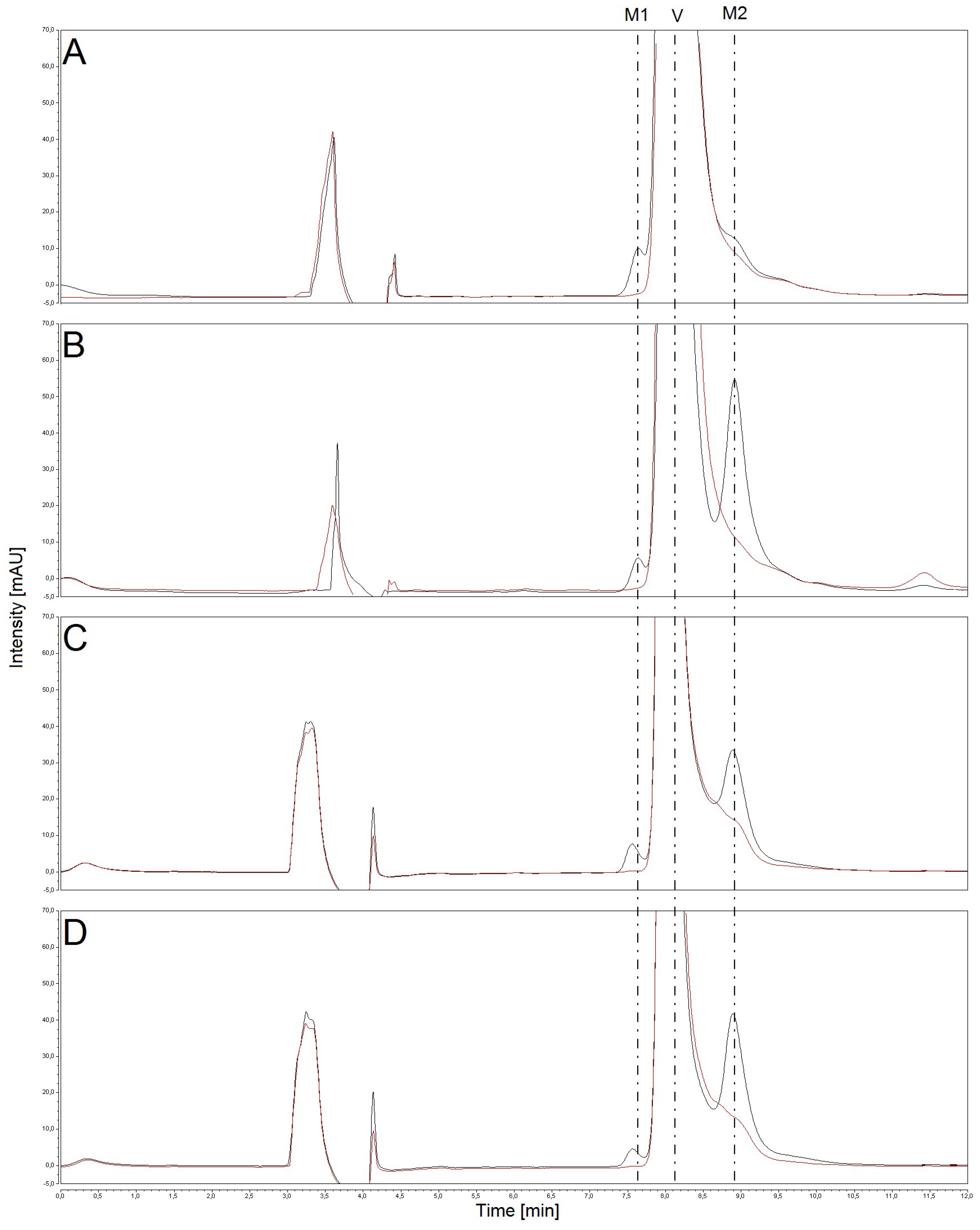
2.1. Oxidation of Vandetanib by Human, Rat, Mouse and Rabbit Hepatic Microsomes
2.2. The Effects of CYP and FMO Enzyme Inhibitors on Vandetanib Oxidation in Human Liver Microsomes
2.3. Correlation of CYP- and FMO-Linked Enzyme Activities in Human Hepatic Microsomes with Vandetanib Oxidation to N-Desmethylvandetanib and Vandetanib-N-Oxide
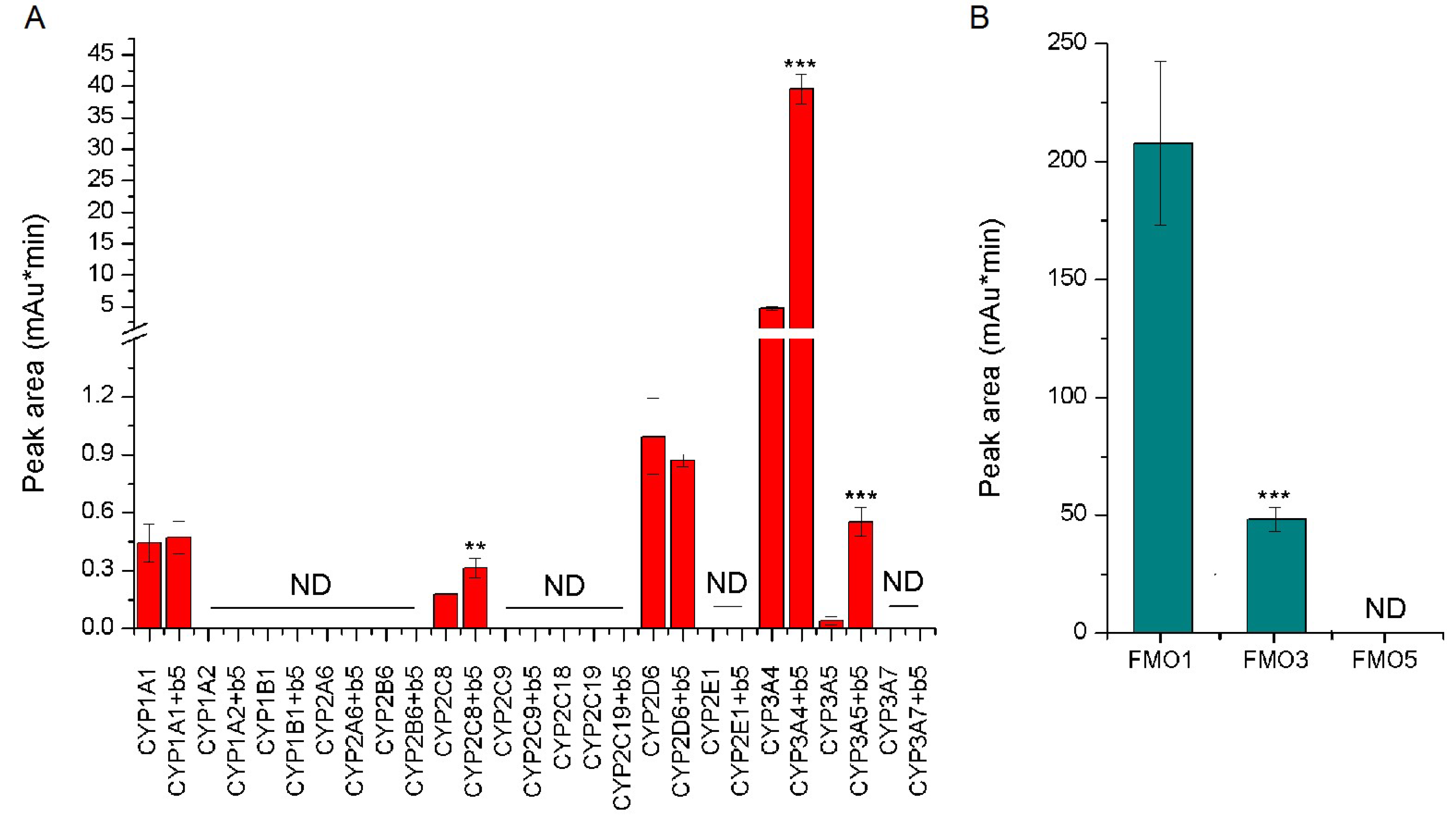
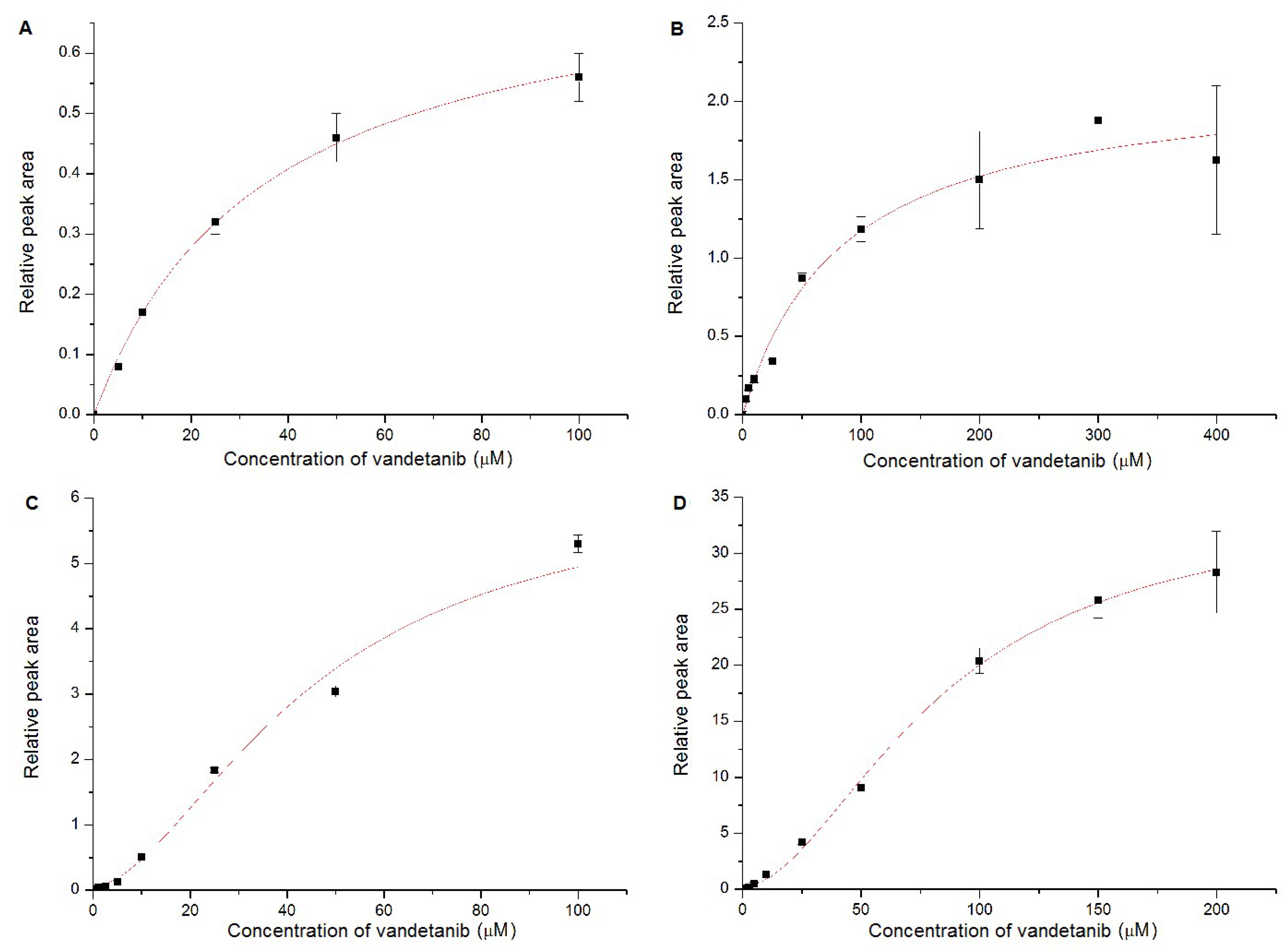
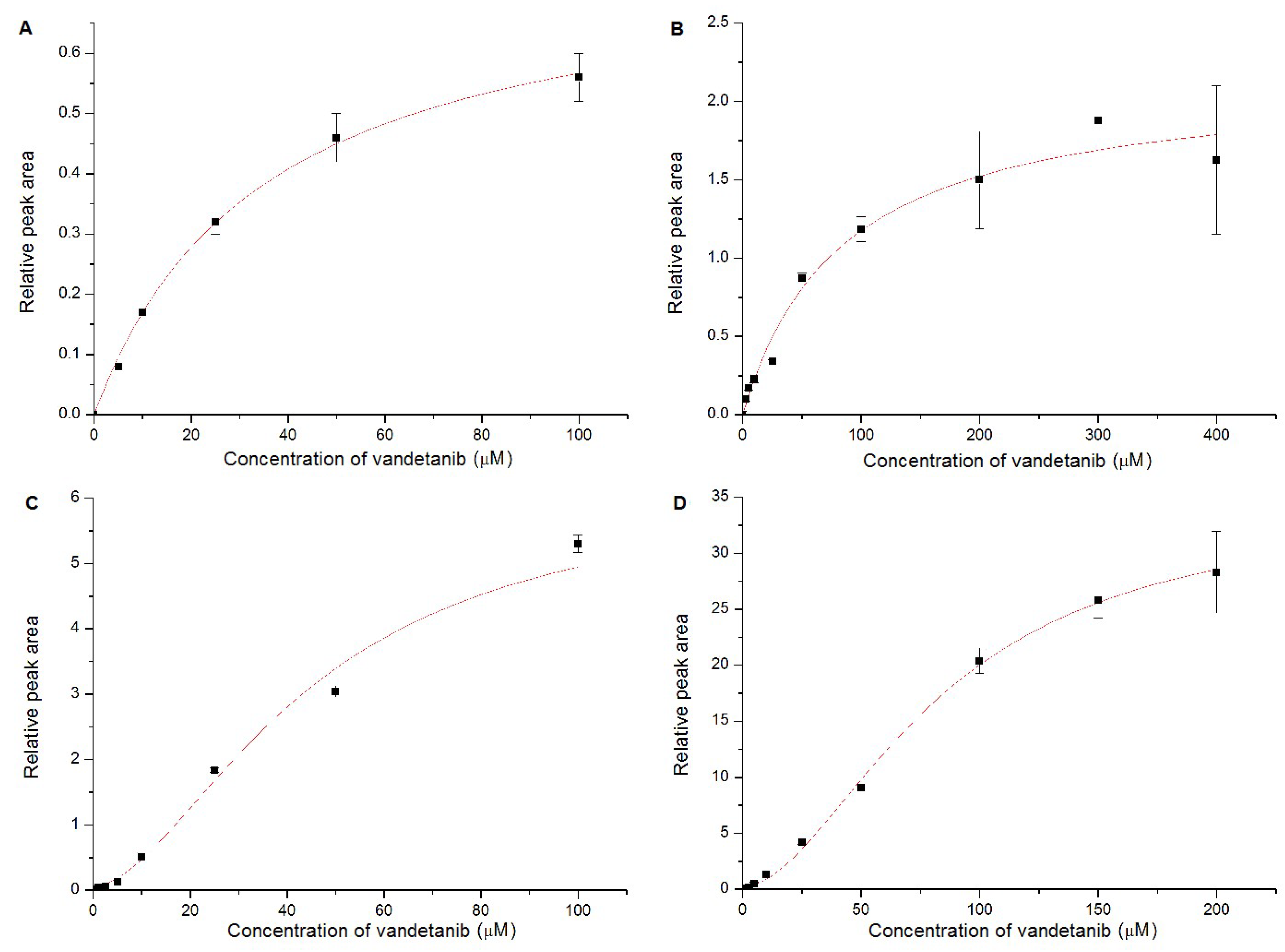
2.4. Oxidation of Vandetanib by Human Recombinant CYP and FMO Enzymes
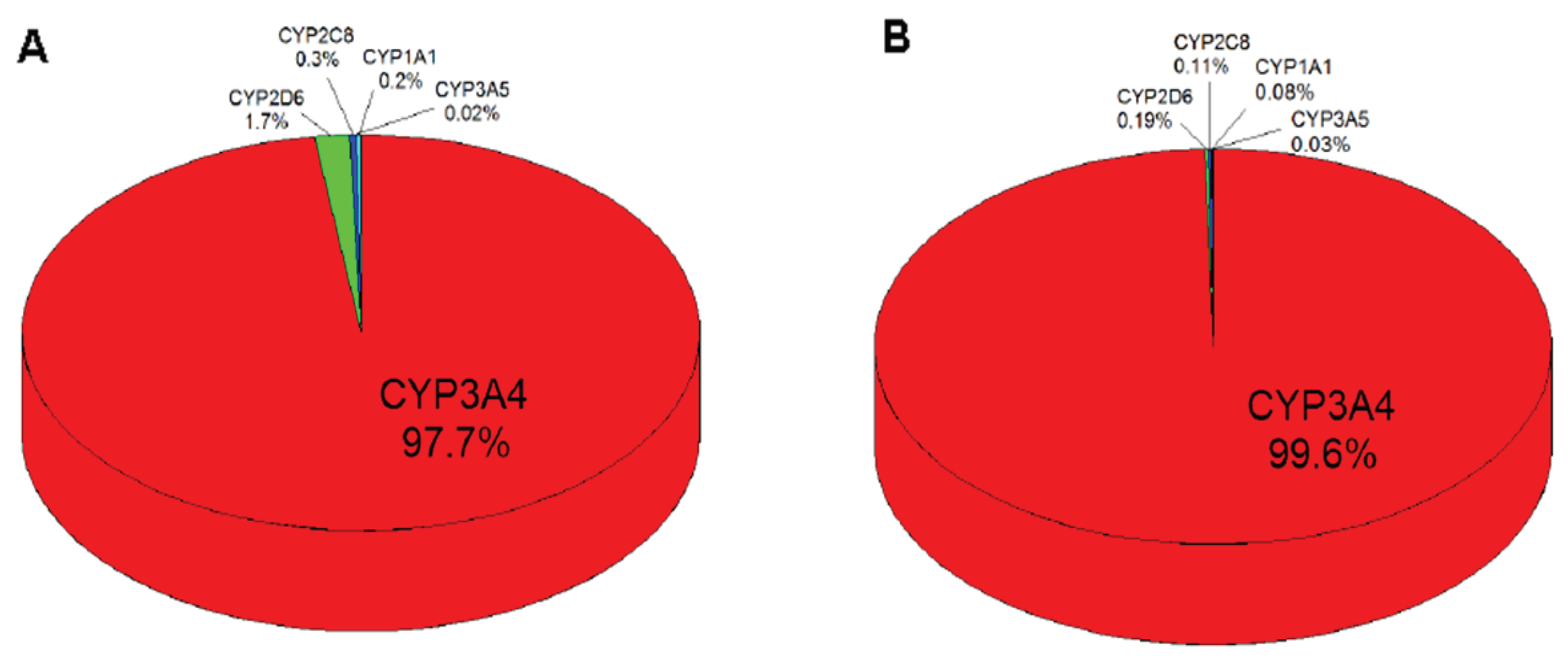
2.5. Contributions of Individual CYPs to Oxidation of Vandetanib to N-Desmethylvandetanib in Human Livers
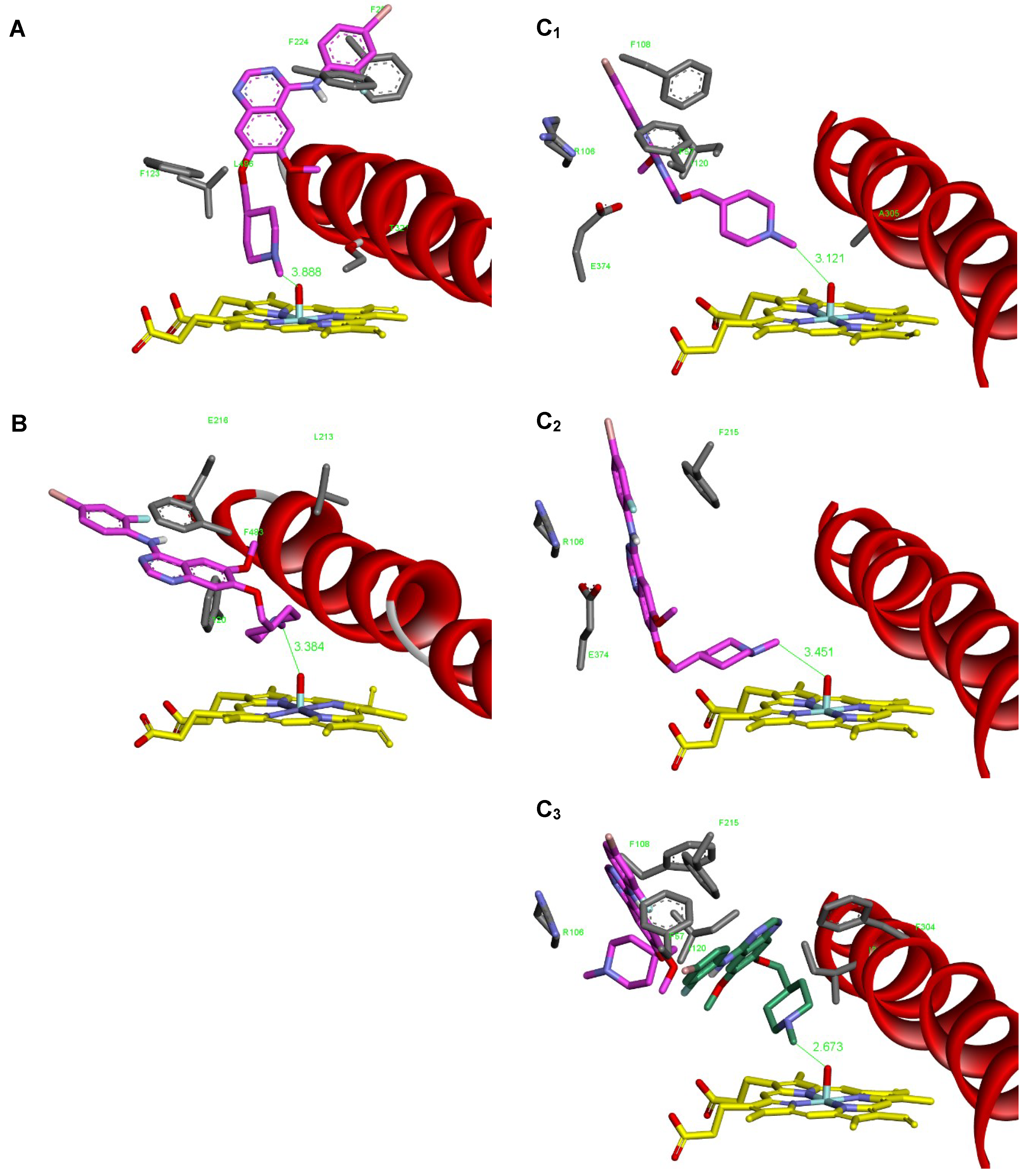
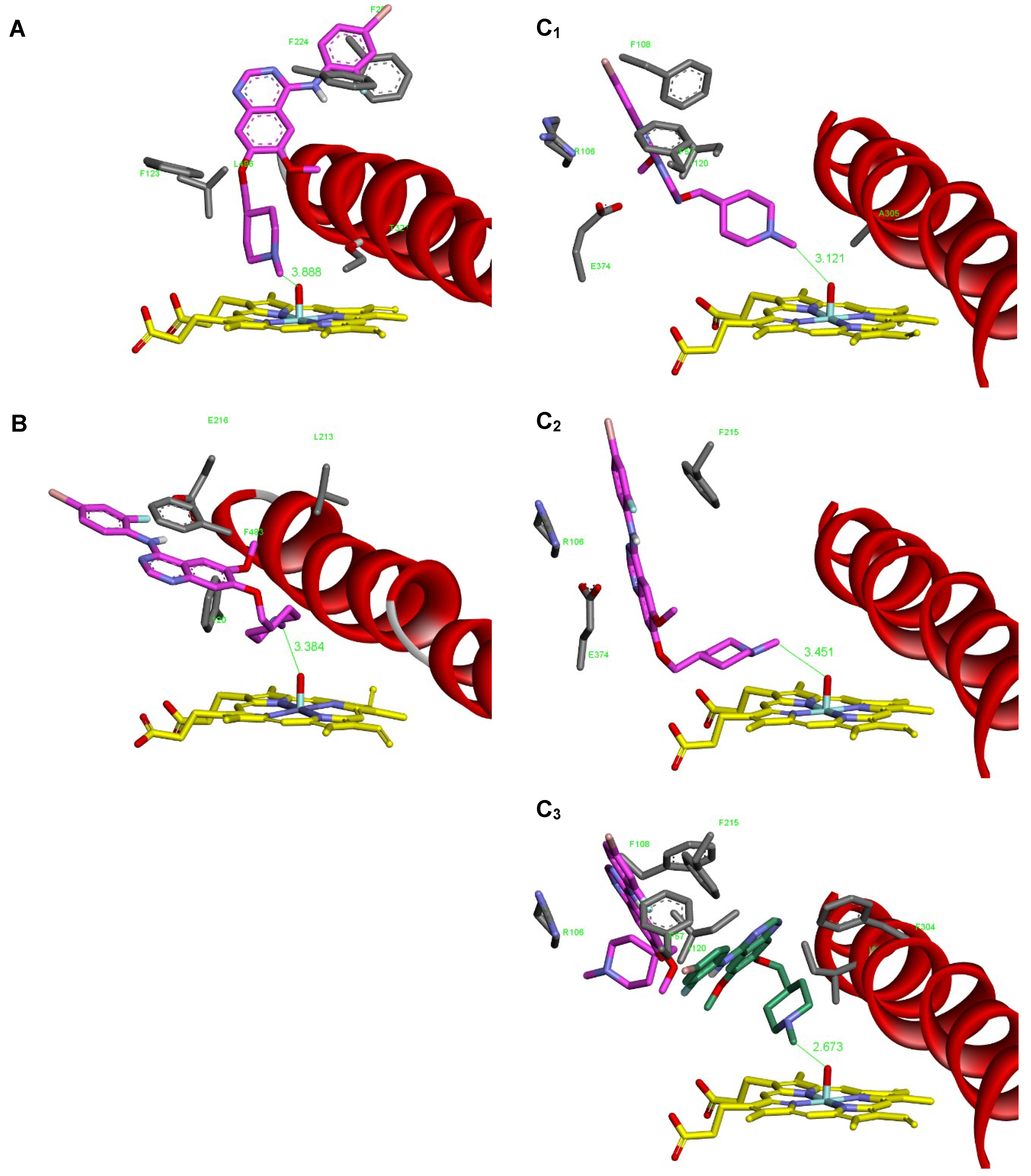
2.6. Binding Of Vandetanib to the Active Site of Compound I of Human CYP1A1, CYP2D6 and CYP3A4
3. Discussion
4. Materials and Methods
4.1. Chemicals and Material
4.2. Oxidation of Vandetanib by Hepatic Microsomes and CYP Enzymes
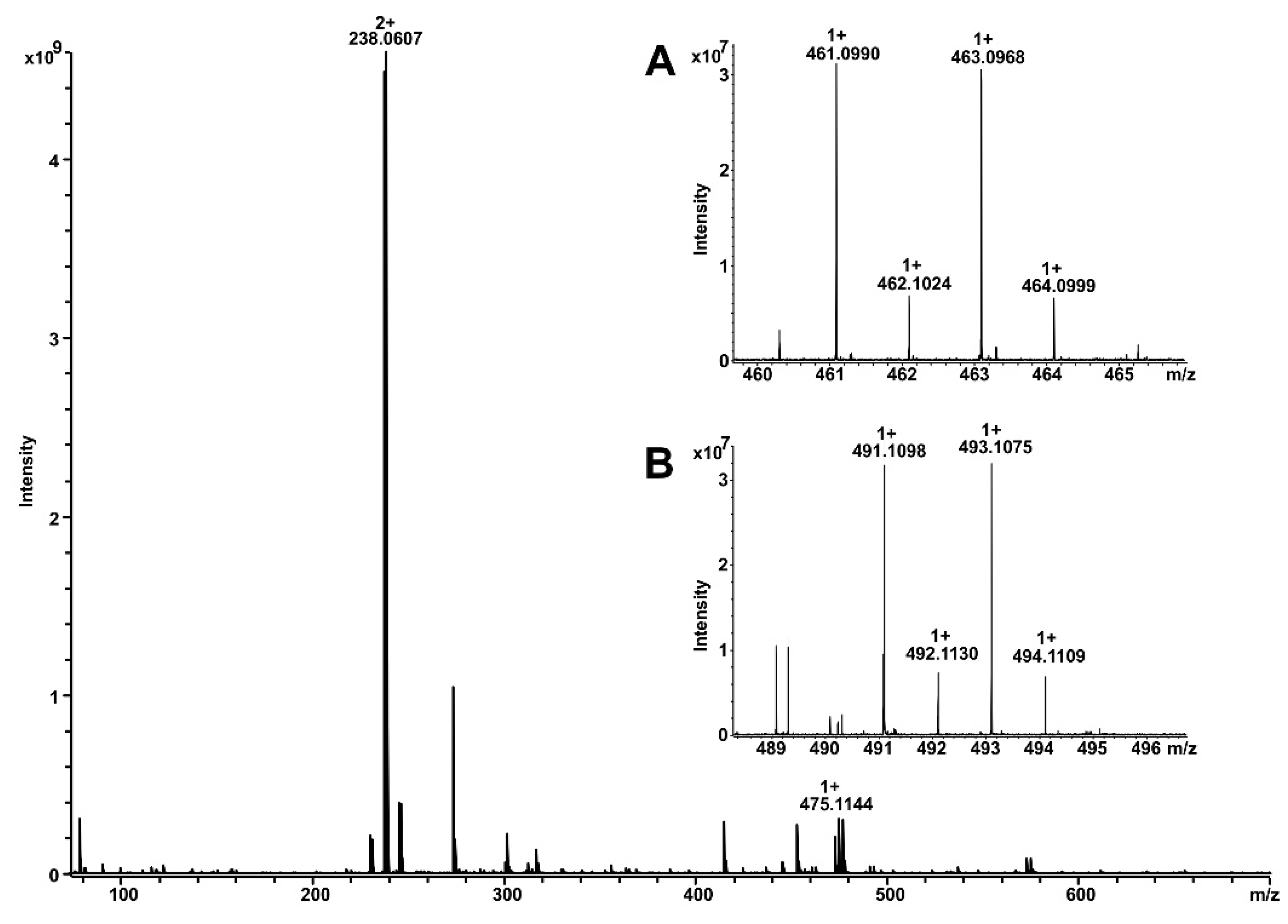
4.3. Identification of Vandetanib Metabolites by Mass Spectrometry
4.4. Inhibition Studies
4.5. Contributions of CYP Enzymes to N-Demethylation of Vandetanib to N-Desmethylvandetanibin in Human Livers
4.6. Molecular Docking of Vandetanib into Compounds I of Human CYP1A1, 2D6 and 3A4
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-NF | α-naphthoflavone |
| CYP | cytochrome P450 |
| DDTC | diethyldithiocarbamate |
| DMSO | dimethyl sulfoxide |
| EGFR | epidermal growth factor receptor |
| FMO | flavin-containing monooxygenase |
| HPLC | high-performance liquid chromatography |
| POR | NADPH:CYP oxidoreductase |
| RET | rearranged during transfection |
| r.t. | retention time |
| TIE2 | tyrosine kinase with immunoglobulin and EGF domains-2 |
| TK | tyrosine kinase |
| TKI | tyrosine kinase inhibitor |
| VEGFR-2 | vascular endothelial growth factor receptor |
References
- Heger, Z.; Cernei, N.; Kudr, J.; Gumulec, J.; Blazkova, I.; Zitka, O.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. A novel insight into the cardiotoxicity of antineoplastic drug doxorubicin. Int. J. Mol. Sci. 2013, 14, 21629–21646. [Google Scholar] [CrossRef] [PubMed]
- Heger, Z.; Skalickova, S.; Zitka, O.; Adam, V.; Kizek, R. Apoferritin applications in nanomedicine. Nanomedicine (Lond) 2014, 9, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Reibenwein, J.; Krainer, M. Targeting signaling pathways in ovarian cancer. Expert Opin. Ther. Targets 2008, 12, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors—a review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, L.F.; Stokem, E.S.; Thomas, A.P.; Johnstone, C.; Plé, P.A.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Kendrew, J.; Curwen, J.O. Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J. Med. Chem. 2002, 45, 1300–1312. [Google Scholar] [CrossRef]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar]
- Ciardiello, F.; Valuto, R.; Damiano, V.; Valuto, R.; Trojanu, T.; Vitaglian, D.; Carlomagno, F.; Veneziani, B.M.; Fontanini, G.; Bianco, A.R.; et al. Antitumor effects of ZD6474, a small molecule vascular endothelial growth factor receptor tyrosine kinase inhibitor, with additional activity against epidermal growth factor receptor tyrosine kinase. Clin. Cancer Res. 2003, 9, 1546–1556. [Google Scholar]
- Commander, H.; Whiteside, G.; Perry, C. Vandetanib: first global approval. Drugs 2011, 71, 1355–1365. [Google Scholar] [CrossRef]
- Martin, P.; Oliver, S.; Robertson, J.; Kennedy, S.J.; Read, J.; Duvauchelle, T. Pharmacokinetic drug interactions with vandetanib during coadministration with rifampicin or itraconazole. Drugs R. D. 2011, 11, 37–51. [Google Scholar] [CrossRef]
- Vozniak, J.M.; Jacobs, J.M. Vandetanib. J. Adv. Pract. Oncol. 2012, 3, 112–116. [Google Scholar] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Wells, A. EGF receptor. Int. J. Biochem. Cell. Biol. 1999, 31, 637–643. [Google Scholar] [CrossRef]
- Kowanetz, M.; Ferrara, N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin. Cancer Res. 2006, 12, 5018–5022. [Google Scholar] [CrossRef] [PubMed]
- Dvořáková, S.; Václavíková, E.; Sýkorová, V.; Včelák, J.; Novák, Z.; Dušková, J.; Ryska, A.; Laco, J.; Čáp, J.; Kodetová, D.; et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinomas. Mol. Cell. Endocrinol. 2008, 284, 21–27. [Google Scholar]
- Václavíková, E.; Dvořáková, S.; Sýkorová, V.; Bilek, R.; Dvořáková, K.; Vlček, P.; Skaba, R.; Zelinka, T.; Bendlová, B. RET mutation Tyr791Phe: the genetic cause of different diseases derived from neural crest. Endocrine 2009, 36, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Pacini, F.; Tuttle, R.M.; Schlumberger, M. Management of advanced medullary thyroid cancer. Lancet Diabetes Endocrinol. 2016, 4, 64–71. [Google Scholar] [CrossRef]
- Hadoux, J.; Schlumberger, M. Chemotherapy and tyrosine-kinase inhibitors for medullary thyroid cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 335–347. [Google Scholar] [CrossRef]
- FDA Center for Drug Evaluation and Research. Approval Package for Vandetanib, Application Number NDA 022405Orig1s000; Medical Review. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022405Orig1s000MedR.pdf (accessed on 19 May 2011).
- Martin, P.; Oliver, S.; Kennedy, S.J.; Partridge, E.; Hutchison, M.; Clarke, D.; Gilda, P. Pharmacokinetics of vandetanib: three phase I studies in healthy subjects. Clin. Ther. 2012, 34, 221–237. [Google Scholar] [CrossRef]
- Fujita, K. Cytochrome P450 and anticancer drugs. Curr. Drug Metab. 2006, 7, 23–37. [Google Scholar] [CrossRef]
- Bates, D. ZD-6474. AstraZeneca. Curr. Opin. Investig. Drugs 2013, 4, 1468–1472. [Google Scholar]
- Australian Public Assessment Report for vandetanib. Caprelsa Vandetanib; AstraZeneca Pty Ltd PM-2011-03002-3-4; Therapeutic Goods Administration: Canberra, Australia, 2013; pp. 1–73.
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Amer, S.M.; Al-Shakliah, N.S. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC-ESI-MS/MS. Chem. Cent. J. 2018, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Nakano, M.; Omak, Y.; Ueng, Y.F.; Guengerich, F.P.; Shimada, T. Roles of cytochrome b5 in the oxidation of testosterone and nifedipine by recombinant cytochrome P450 3A4 and by human liver microsomes. Arch. Biochem. Biophys. 1996, 325, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Shimada, T.; Martin, M.V.; Guengerich, F.P. Stimulation of cytochrome P450 reactions by apo-cytochrome b5: evidence against transfer of heme from cytochrome P450 3A4 to apo-cytochrome b5 or heme oxygenase. J. Biol. Chem. 2001, 276, 30885–30891. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Indra, R.; Mizerovská, M.; Cerná, V.; Rupertová, M.; Martínek, V.; Eckschlager, T.; Kizek, R.; Frei, E. Cytochrome b5 increases cytochrome P450 3A4-mediated activation of anticancer drug ellipticine to 13-hydroxyellipticine whose covalent binding to DNA is elevated by sulfotransferases and N,O-acetyltransferases. Chem. Res. Toxicol. 2017, 25, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Indra, R.; Frei, E.; Kopečková, K.; Schmeiser, H.H.; Eckschlager, T.; Adam, V.; Heger, Z.; Arlt, V.M.; Martínek, V. Cytochrome b5 plays a dual role in the reaction cycle of cytochrome P450 3A4 during oxidation of the anticancer drug ellipticine. Monatsh. Chem. 2017, 148, 1983–1991. [Google Scholar]
- Guengerich, F.P. Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Ballard, J.E.; Prueksaritanont, T.; Tang, C. Hepatic metabolism of MK-0457, a potent aurora kinase inhibitor: Interspecies comparison and role of human cytochrome P450 and flavin-containing monooxygenase. Drug Metab. Dispos. 2007, 9, 1447–1451. [Google Scholar] [CrossRef]
- Gao, C.; Catucci, G.; Gilardi, G.; Sadeghi, S.J. Binding of methimazole and NADP(H) to human FMO3: In vitro and in silico studies. Int. J. Biol. Macromol. 2018, 118, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Porter, T.D. The roles of cytochrome b5 in cytochrome P450 reactions. J. Biochem. Mol. Toxicol. 2002, 16, 311–316. [Google Scholar] [CrossRef]
- Schenkman, J.B.; Jansson, I. The many roles of cytochrome b5. Pharmacol. Ther. 2003, 97, 139–152. [Google Scholar] [CrossRef]
- McLaughin, L.A.; Ronseaux, S.; Finn, R.D.; Henderson, C.J.; Wolf, C.R. Deletion of microsomal cytochrome b5 profoundly affects hepatic and extrahepatic drug metabolism. Mol. Pharmacol. 2010, 75, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Indra, R.; Mizerovská, M.; Frei, E.; Schmeiser, H.H.; Kopka, K.; Philips, D.H.; Arlt, V.M. NADH:Cytochrome b5 reductase and cytochrome b5 can act as sole electron donors to human cytochrome P450 1A1-mediated oxidation and DNA adduct formation by benzo[a]pyrene. Chem. Res. Toxicol. 2016, 29, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Moserova, M.; Mrizova, I.; Dracinska, H.; Martinek, V.; Indra, R.; Frei, E.; Adam, V.; Kizek, R.; Schmeiser, H.H.; et al. Induced expression of microsomal cytochrome b5 determined at mRNA and protein levels in rats exposed to ellipticine, benzo[a]pyrene, and 1-phenylazo-2-naphthol (Sudan I). Monatsh. Chem. 2016, 147, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.; Mrizova, I.; Barta, F.; Indra, R.; Moserova, M.; Kopka, K.; Schmeiser, H.H.; Wolf, C.R.; Henderson, C.J.; Stiborova, M.; et al. Cytochrome b5 impacts on cytochrome P450-mediated metabolism of benzo[a]pyrene and its DNA adduct formation: studies in hepatic cytochrome b5/P450 reductase null (HBRN) mice. Arch. Toxicol. 2018, 92, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Kimura, T.; Yokoi, T.; Itoh, S.; Kamataki, T. Rat liver flavin-containing monooxygenase (FMO): cDNA cloning and expression in yeast. Biochim. Biophys. Acta 1993, 1173, 165–171. [Google Scholar] [CrossRef]
- Krueger, S.K.; Williams, D.E. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 2005, 106, 357–387. [Google Scholar] [CrossRef] [Green Version]
- Rendic, S.; DiCarlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–480. [Google Scholar] [CrossRef]
- Nedelcheva, V.; Gut, I. P450 in the rat and man: methods of investigation, substrate specificities and relevance to cancer. Xenobiotica 1994, 24, 1151–1175. [Google Scholar] [CrossRef]
- Stiborová, M.; Martínek, V.; Rýdlová, H.; Hodek, P.; Frei, E. Sudan I is a potential carcinogen for humans: evidence for its metabolic activation and detoxication by human recombinant cytochrome P450 1A1 and liver microsomes. Cancer Res. 2002, 62, 5678–5684. [Google Scholar]
- Stiborová, M.; Martínek, V.; Rýdlová, H.; Koblas, T.; Hodek, P. Expression of cytochrome P450 1A1 and its contribution to oxidation of a potential human carcinogen 1-phenylazo-2-naphthol (Sudan I) in human livers. Cancer Lett. 2005, 220, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Shimizu, M.; Nagashima, T.; Minoshima, M.; Murayama, N. Rat cytochrome P450 2C11 in liver microsomes involved in oxidation of anesthetic agent propofol and deactivated by prior treatment with propofol. Drug Metab. Dispos. 2006, 34, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Zuber, R.; Modrianský, M.; Dvorák, Z.; Rohovský, P.; Ulrichová, J.; Simánek, V.; Anzenbacher, P. Effect of silybin and its congeners on human liver microsomal cytochrome P450 activities. Phytother. Res. 2002, 16, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Moskaleva, N.; Moysa, A.; Novikova, S.; Tikhonova, O.; Zgoda, V.; Archakov, A. Spaceflight effects on cytochrome P450 content in mouse liver. PLoS One 2015, 10, e0142374. [Google Scholar] [CrossRef] [PubMed]
- Kopečková, K.; (Department of Oncology, 2nd Medical Faculty, Charles University and University Hospital Motol, Prague 5, Czech Republic). Personal Communication, 2019.
- Williams, D.E.; Hale, S.E.; Muerhoff, A.S.; Masters, B.S. Rabbit lung flavin-containing monooxygenase. Purification, characterization, and induction during pregnancy. Mol. Pharmacol. 1985, 28, 381–390. [Google Scholar]
- Dannan, G.A.; Guengerich, F.P.; Waxman, D.J. Hormonal regulation of rat liver microsomal enzymes. Role of gonadal steroids in programming, maintenance, and suppression of delta 4-steroid 5 alpha-reductase, flavin-containing monooxygenase, and sex-specific cytochromes P-450. J. Biol. Chem. 1986, 261, 10728–10735. [Google Scholar]
- Tynes, R.E.; Philpot, R.M. Tissue- and species-dependent expression of multiple forms of mammalian microsomal flavin-containing monooxygenase. Mol. Pharmacol. 1987, 31, 569–574. [Google Scholar]
- Fumarola, A.; Di Fjord, A.; Dainelli, M.; Grani, G.; Calvanese, A. Medical treatment of hyperthyroidism: state of the art. Exp. Clin. Endocrinol. Diabetes. 2010, 118, 678–684. [Google Scholar] [CrossRef]
- Stiborová, M.; Stiborová-Rupertová, M.; Bořek-Dohalská, L.; Wiessler, M.; Frei, E. Rat microsomes activating the anticancer drug ellipticine to species covalently binding to deoxyguanosine in DNA are a suitable model mimicking ellipticine bioactivation in humans. Chem. Res. Toxicol. 2003, 16, 38–47. [Google Scholar] [CrossRef]
- Hodek, P.; Koblihova, J.; Kizek, R.; Frei, E.; Arlt, V.M.; Stiborova, M. The relationship between DNA adduct formation by benzo[a]pyrene and expression of its activation enzyme cytochrome P450 1A1 in rats. Environ. Toxicol. Pharmacol. 2013, 36, 989–996. [Google Scholar] [CrossRef]
- Wiechelman, K.J.; Braun, R.D.; Fitzpatrick, J.D. Investigation of the bicinchoninic acid protein assay: identified cation of the groups responsible for color formation. Anal. Biochem. 1988, 175, 231–237. [Google Scholar] [CrossRef]
- Kotrbova, V.; Mrazova, B.; Moserova, M.; Martinek, V.; Hodek, P.; Hudecek, J.; Frei, E.; Stiborova, M. Cytochrome b5 shifts oxidation of the anticancer drug ellipticine by cytochromes P450 1A1 and 1A2 from its detoxication to activation, thereby modulating its pharmacological efficacy. Biochem. Pharmacol. 2011, 82, 669–680. [Google Scholar] [CrossRef]
- Stiborová, M.; Poljaková, J.; Martínková, E.; Ulrichová, J.; Simánek, V.; Dvořák, Z.; Frei, E. Ellipticine oxidation and DNA adduct formation in human hepatocytes is catalyzed by human cytochromes P450 and enhanced by cytochrome b5. Toxicology 2012, 302, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Šulc, M.; Indra, R.; Moserová, M.; Schmeiser, H.H.; Frei, E.; Arlt, V.M.; Stiborová, M. The impact of individual cytochrome P450 enzymes on oxidative metabolism of benzo[a]pyrene in human livers. Environ. Mol. Mutagen. 2016, 57, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Levová, K.; Bárta, F.; Shi, Z.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Arlt, V.M. Bioactivation versus detoxication of the urothelial carcinogen aristolochic acid I by human cytochrome P450 1A1 and 1A2. Toxicol. Sci. 2012, 125, 345–358. [Google Scholar] [CrossRef]
- Stiborová, M.; Borek-Dohalska, L.; Aimova, D.; Kotrbova, V.; Kukackova, K.; Janouchova, K.; Rupertova, M.; Ryslava, H.; Hudecek, J.; Frei, E. Oxidation pattern of the anticancer drug ellipticine by hepatic microsomes—similarity between human and rat systems. Gen. Physiol. Biophys. 2006, 25, 245–261. [Google Scholar] [PubMed]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef]
- Wang, A.; Savas, U.; Hsu, M.H.; Stout, C.D.; Johnson, E.F. Contributions of ionic interactions and protein dynamics to cytochrome P450 2D6 (CYP2D6) substrate and inhibitor binding. J. Biol. Chem. 2015, 290, 5092–5104. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Choti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Samuels, E.R.; Sevrioukova, I. Inhibition of human CYP3A4 by rationally designed ritonavir-like compounds: Impact and interplay of the side group functionalities. Mol. Pharm. 2018, 15, 279–288. [Google Scholar] [CrossRef]
- Pontikis, G.; Borden, J.; Martínek, V.; Florián, J. Linear energy relationships for the octahedral preference of Mg, Ca and transition metal ions. J. Phys. Chem. 2009, 113, 3588–3593. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision A. 02. Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Martínek, V.; Bárta, F.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. Comparison of the oxidation of carcinogenic aristolochic acid I and II by microsomal cytochromes P450 in vitro: Experimental and theoretical approaches. Monatsh. Chem. 2017, 148, 1971–1981. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Inhibitor a | % of Inhibition | IC50 (μM) |
|---|---|---|---|
| N-Desmethyvandetanib Formation | |||
| CYP | α-Napththoflavone (CYP1A) | 0 | NA b |
| Diamantane (CYP2B) | 0 | NA | |
| Sulfaphenazole (CYP2C) | 38 ± 4 *** | NA | |
| Quinidine (CYP2D) | 20 ± 4 ** | NA | |
| DDTC (CYP2A, CYP2E1) | 0 | NA | |
| Ketoconazole (CYP3A) | 98 ± 3 *** | 2 ± 0.2 | |
| Vandetanib-N-oxide Formation | |||
| FMO | Methimazol (FMOs) | 79 ± 4 *** | 6 ± 0.5 |
| a No. | b Total CYPs | c POR activity | d Cyt b5 | e CYP1A2 | e CYP2A6 | e CYP2B6 | e CYP2C8 | e CYP2C9 | e CYP2C19 | e CYP2D6 | e CYP2E1 | e CYP3A4 | e CYP4A11 | e FMO | f M1 | f M2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HG03 | 290 | 450 | 380 | 170 | 2000 | 51 | 200 | 1700 | 44 | 110 | 1800 | 6100 | 1600 | g NM | 3.639 | h ND |
| HG103 | 340 | 210 | 790 | 310 | 440 | 7.2 | 39 | 2300 | 23 | 65 | 1100 | 2200 | 1600 | 1400 | 1.021 | 0.004 |
| HG24 | 260 | 260 | 550 | 1700 | 1500 | 35 | 190 | 3000 | 41 | NM | 2300 | 4000 | 1800 | 1500 | 1.698 | 0.004 |
| HG32 | 170 | 330 | 580 | 730 | 520 | 0.68 | 20 | 450 | 4.8 | 46 | 1200 | 2000 | 680 | 920 | 0.870 | ND |
| HG42 | 670 | 510 | 500 | 700 | 2200 | 150 | 480 | 1600 | 7,4 | 95 | 1600 | 15000 | 1400 | 2000 | 8.887 | 0.015 |
| HG43 | 270 | 210 | 640 | 580 | 770 | 14 | 25 | 1800 | 440 | 4 | 780 | 4600 | 1800 | 920 | 1.830 | ND |
| HG74 | 220 | 200 | 600 | 520 | 360 | 13 | 130 | 2100 | 55 | 120 | 1400 | 2700 | 1300 | 1200 | 0.714 | 0.004 |
| HG93 | 430 | 320 | 450 | 691 | 350 | 43 | 270 | 2200 | 75 | 49 | 2800 | 2800 | 1800 | 3500 | 0.966 | 0.112 |
| HK23 | 380 | 380 | 700 | 960 | 1100 | 24 | 160 | 2100 | 110 | 140 | 2100 | 6800 | 780 | 2500 | 4.137 | 0.008 |
| HK27 | 300 | 450 | 730 | 1320 | 1320 | 31 | 180 | 480 | 460 | 130 | 3000 | 4910 | 1110 | 2230 | 1.762 | ND |
| HK31 | 580 | 540 | 770 | 1220 | 2160 | 8.1 | 130 | 1690 | 172 | 3,4 | 1660 | 8210 | 2010 | 3020 | 4.124 | 0.015 |
| HK34 | 500 | 460 | 890 | 1000 | 1500 | 39 | 220 | 1900 | 45 | 100 | 6000 | 5200 | 1100 | 2700 | 3.193 | ND |
| Total CYPs | POR | Cyt b5 | CYP1A2 | CYP2A6 | CYP2B6 | CYP2C8 | CYP2C9 | CYP2C19 | CYP2D6 | CYP2E1 | CYP3A4 | CYP4A11 | FMO | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a M1 | 0.786 ** | 0.700 | −0.114 | 0.018 | 0.778 ** | 0.826 *** | 0.747 ** | −0.034 | −0.168 | 0.165 | 0.043 | 0.984 *** | 0.002 | 0.270 |
| a M2 | 0.166 | 0.035 | −0.5743 | −0.157 | −0.339 | 0.093 | 0.288 | −0.039 | 0.088 | −0.343 | 0.714 ** | −0.200 | 0.315 | 0.736 ** |
| Simulated System | The Most Stable Productive Orientations of the Neutral Form of Vandetanib in the Complex with CYPs | |
|---|---|---|
| Estimated Free Energy of Binding (kcal/mol) | O(Comp I)-N-CH3Distance [Å]a | |
| CYP1A1 (4I8V) | −8.64 | 3.89 |
| CYP2D6 (3TDA) | −10.21 | 3.38 |
| CYP3A4 (1W0E) | −9.96 | 3.12 |
| CYP3A4 (6BD7) | −9.63 | 3.45 |
| CYP3A4 (6BD7) + second vandetanib molecule | −9.30 | 2.67 |
| CYP Enzyme | Kinetical Characteristics | ||
|---|---|---|---|
| a VMax | b K0.5 | c Hill Coefficient | |
| CYP1A1 | 0.76 ± 0.02 | 35.12 ± 2.75 | NA |
| CYP2D6 | 2.16 ± 0.15 | 84.36 ± 18.14 | NA |
| CYP3A4 | 6.17 ± 1.32 | 45.02 ± 13.62 | 1.73 ± 0.56 |
| CYP3A4+cyt b5 | 34.32 ± 1.59 | 83.65 ± 5.55 | 1.83 ± 0.13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indra, R.; Pompach, P.; Martínek, V.; Takácsová, P.; Vavrová, K.; Heger, Z.; Adam, V.; Eckschlager, T.; Kopečková, K.; Arlt, V.M.; et al. Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation. Int. J. Mol. Sci. 2019, 20, 3392. https://doi.org/10.3390/ijms20143392
Indra R, Pompach P, Martínek V, Takácsová P, Vavrová K, Heger Z, Adam V, Eckschlager T, Kopečková K, Arlt VM, et al. Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation. International Journal of Molecular Sciences. 2019; 20(14):3392. https://doi.org/10.3390/ijms20143392
Chicago/Turabian StyleIndra, Radek, Petr Pompach, Václav Martínek, Paulína Takácsová, Katarína Vavrová, Zbyněk Heger, Vojtěch Adam, Tomáš Eckschlager, Kateřina Kopečková, Volker Manfred Arlt, and et al. 2019. "Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation" International Journal of Molecular Sciences 20, no. 14: 3392. https://doi.org/10.3390/ijms20143392
APA StyleIndra, R., Pompach, P., Martínek, V., Takácsová, P., Vavrová, K., Heger, Z., Adam, V., Eckschlager, T., Kopečková, K., Arlt, V. M., & Stiborová, M. (2019). Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation. International Journal of Molecular Sciences, 20(14), 3392. https://doi.org/10.3390/ijms20143392