β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells


 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
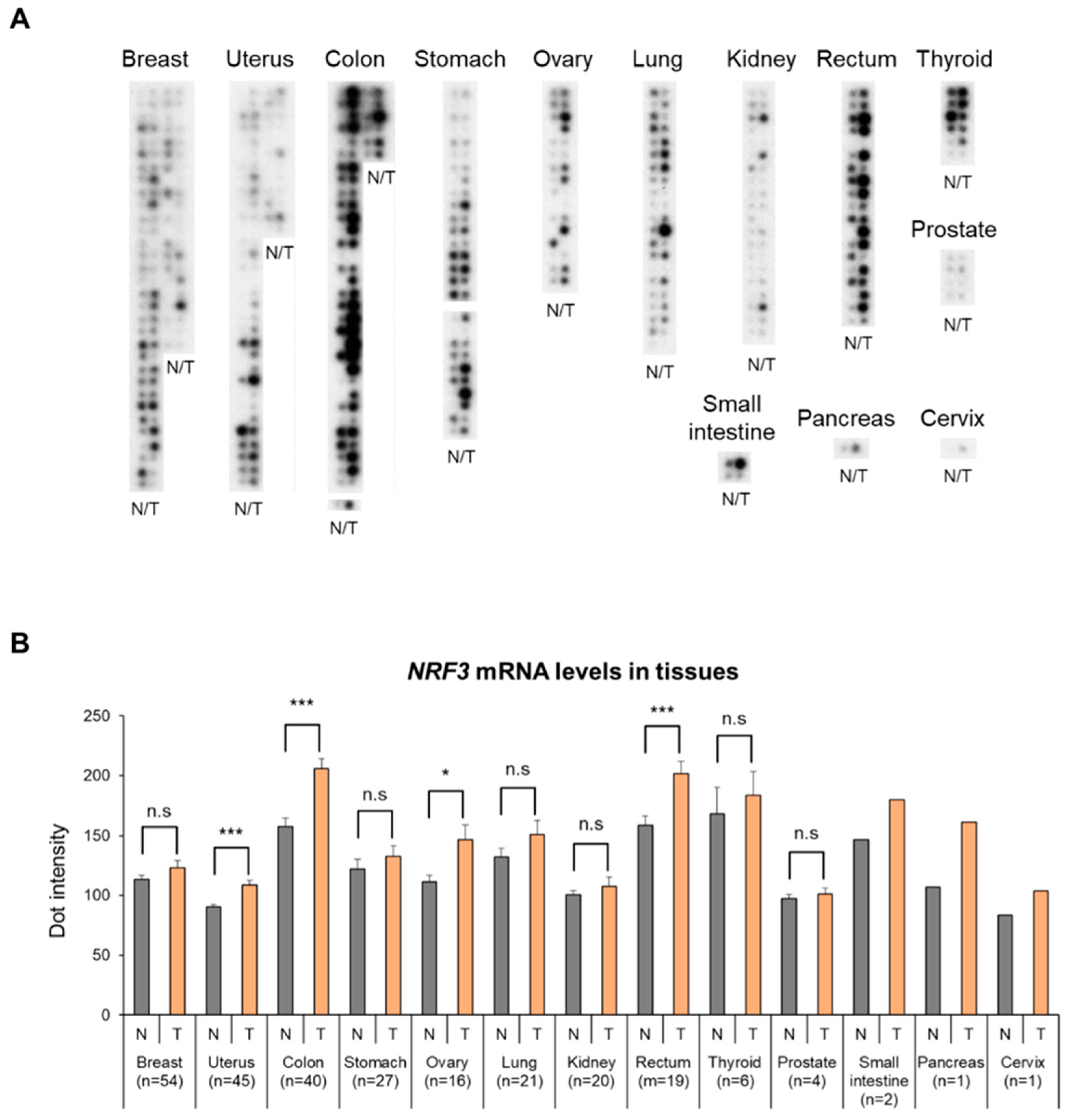
2.1. High Expression of The NRF3 Gene in Human Colorectal Cancer Specimens
2.2. The β-catenin/TCF4 Complex Directly Mediates NRF3 Gene Induction in Cancer Cells
2.3. The β-catenin/TCF4-NRF3 Axis Is Conserved in Other Cancer Cells
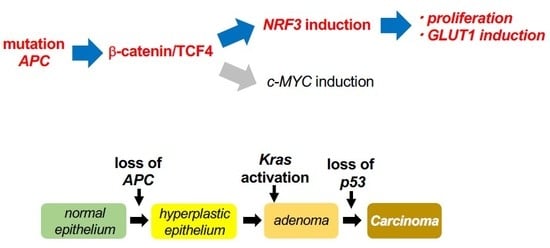
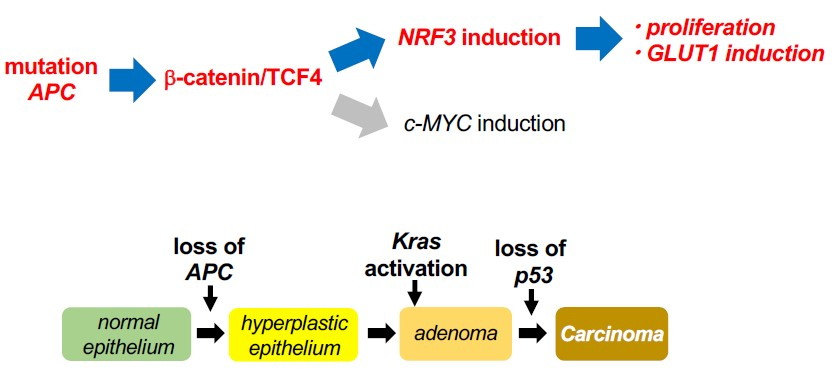
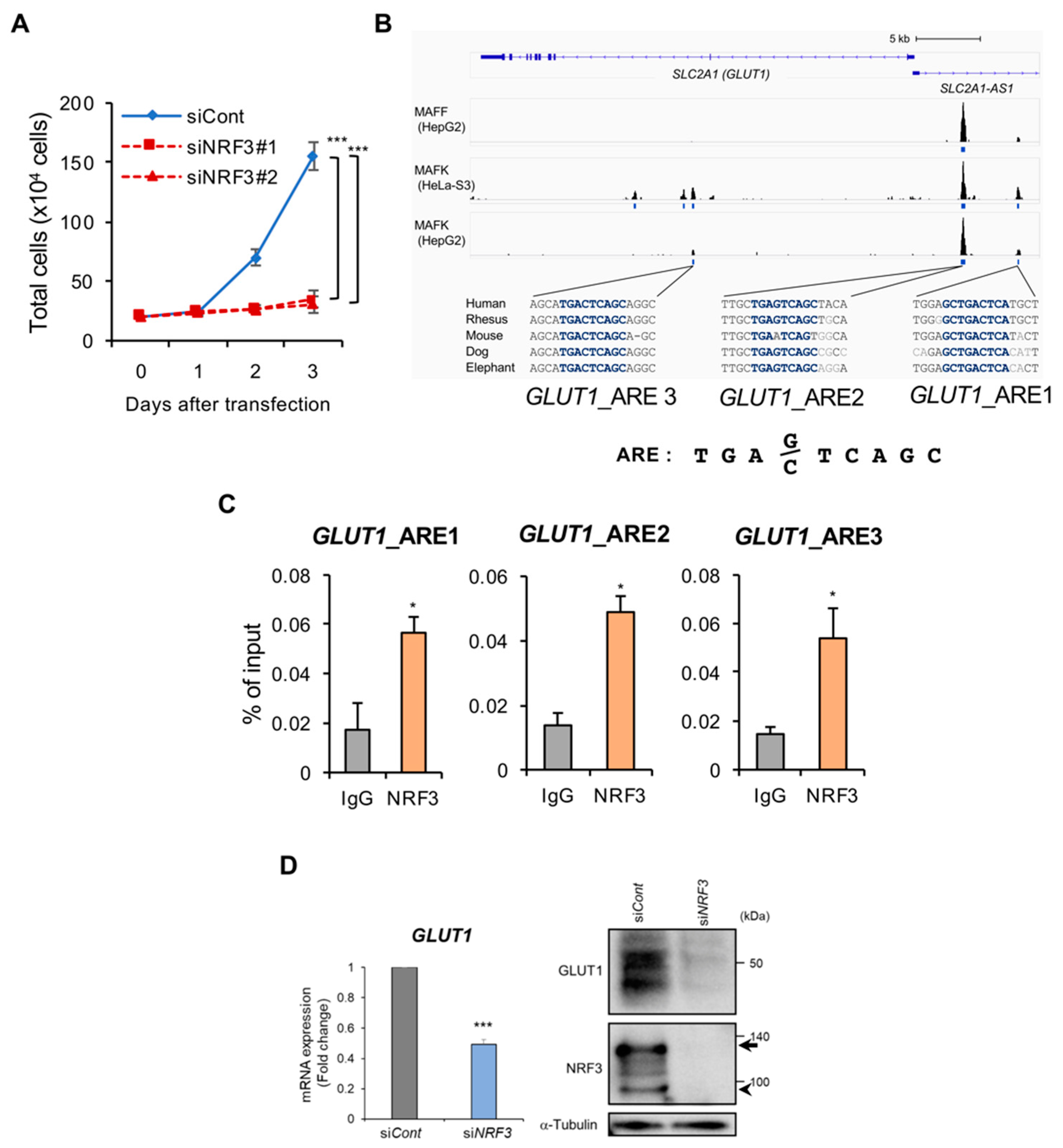
2.4. NRF3 Activates Cell Proliferation and GLUT1 Gene Expression in Colon Cancer Cells
2.5. The β-catenin/Tcf4-Nrf3 Axis Is Conserved in Mouse Intestinal Tumors
2.6. Correlation of Gene Expression between NRF3 and Target Genes of The β-catenin/TCF4 Complex in a Human Cancer Database
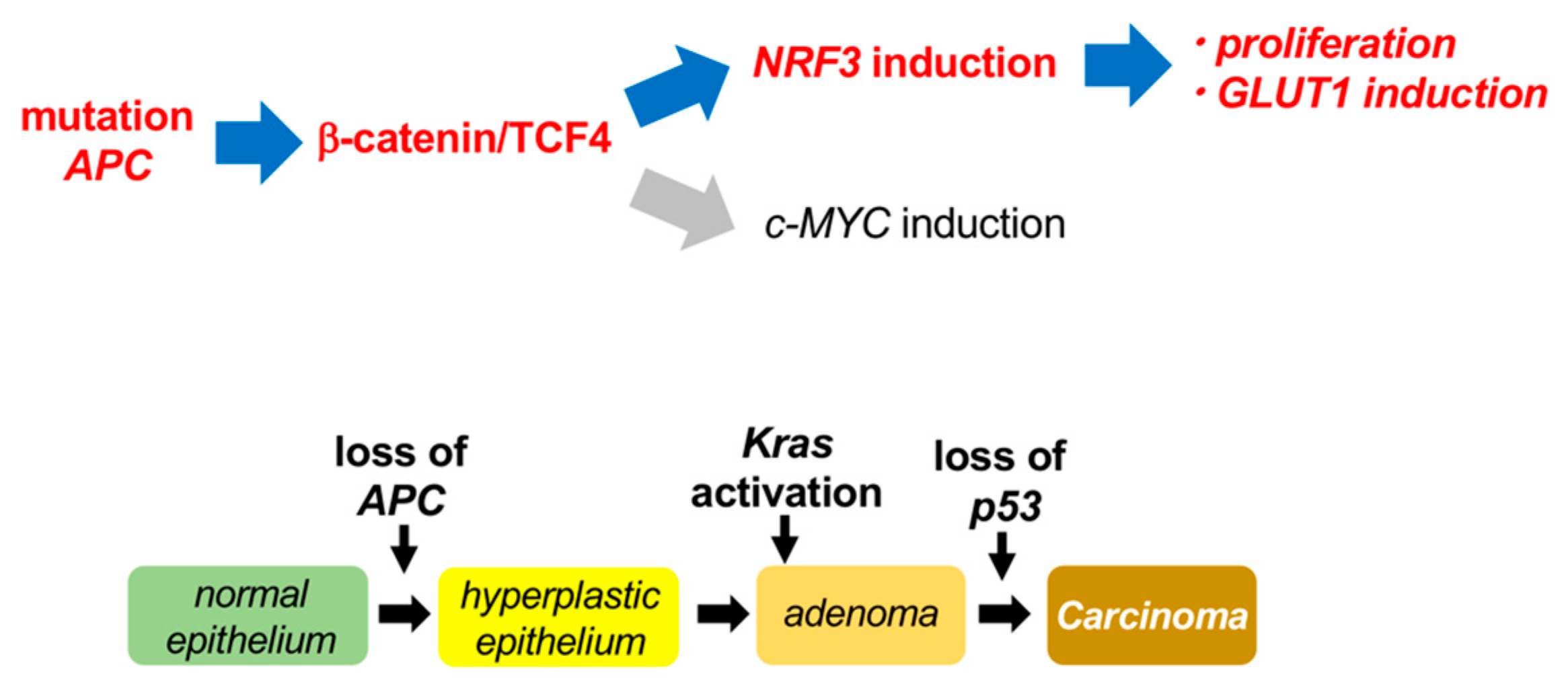
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. RNA Dot Blot Analysis
4.3. Cell Culture
4.4. Apc flox/flox and ApcΔ716 Mice
4.5. Preparation of, Culture of and Apc Gene Deletion in Intestine Organoids Derived from Apcflox/flox Mice
4.6. Transfection
4.7. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
4.8. Cell Fractionation and Western Blot Analysis
4.9. Chromatin Immunoprecipitation (ChIP)
4.10. Cell Proliferation Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| ARE | Antioxidant response element |
| ChIP | Chromatin immunoprecipitation |
| CNC | Cap “N” collar |
| LEF | Lymphoid enhancer-binding factor |
| NFE2L3 | NFE2-like 3 |
| NRF3 | NFE2-related factor 3 |
| TCF4 | T-cell factor 4 |
| WRE | Wnt response element |
References
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center. Available online: http://firebrowse.org/viewGene.html?gene=NFE2L3. TCGA data version 2016_01_28 (accessed on 30 October 2018).
- Kobayashi, A.; Ito, E.; Toki, T.; Kogame, K.; Takahashi, S.; Igarashi, K.; Hayashi, N.; Yamamoto, M. Molecular Cloning and Functional Characterization of a New Cap’n’Collar Family Transcription Factor Nrf3. J. Biol. Chem. 1999, 274, 6443–6452. [Google Scholar] [CrossRef] [PubMed]
- Bugno, M.; Daniel, M.; Chepelev, N.L.; Willmore, W.G. Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim. Biophys. Acta 2015, 1849, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, X.M.; Kensler, T.W.; Motohashi, H. The Keap1-Nrf2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, G.; Blank, V. NFE2L3 (NRF3): The Cinderella of the Cap’n’Collar transcription factors. Cell. Mol. Life Sci. 2011, 3, 3337–3348. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Ohta, T.; Yamamoto, M.; Ohta, M.; Yamamoto, M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar]
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.Q.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.C.; Blank, V. Complexity of CNC Transcription Factors As Revealed by Gene Targeting of the Nrf3 Locus. Mol. Cell. Biol. 2004, 24, 3286–3294. [Google Scholar] [CrossRef]
- Chevillard, G.; Paquet, M.; Blank, V. Nfe2l3 (Nrf3) deficiency predisposes mice to T-cell lymphoblastic lymphoma. Blood 2011, 117, 2005–2008. [Google Scholar] [CrossRef]
- Chowdhury, A.M.M.A.; Katoh, H.; Hatanaka, A.; Iwanari, H.; Nakamura, N.; Hamakubo, T.; Natsume, T.; Waku, T.; Kobayashi, A. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci. Rep. 2017, 7, 12494. [Google Scholar] [CrossRef]
- Wang, C.; Saji, M.; Justiniano, S.E.; Yusof, A.M.; Zhang, X.; Yu, L.; Fernández, S.; Wakely, P., Jr.; La Perle, K.; Nakanishi, H.; et al. RCAN1-4 is a thyroid cancer growth and metastasis suppressor. J. Clin. Investig. Insight 2017, 2, e90651. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhan, M.; Yang, R.; Shi, Y.; Liu, Q.; Wang, J. Elevated expression of NFE2L3 predicts the poor prognosis of pancreatic cancer patients. Cell Cycle 2018, 17, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.T.; Hsieh, F.J.; Chen, S.W.; Chen, C.L.; Shu, H.F.; Li, H. Clinicopathologic correlation of up-regulated genes identified using cDNA microarray and real-time reverse transcription-PCR in human colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 437–443. [Google Scholar] [CrossRef]
- Almstrup, K.; Ottesen, A.M.; Sonne, S.B.; Hoei-Hansen, C.E.; Leffers, H.; Rajpert-De Meyts, E.; Skakkebaek, N.E. Genomic and gene expression signature of the pre-invasive testicular carcinoma in situ. Cell Tissue Res. 2005, 322, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.K.; Park, S.H.; Jang, Y.K. Molecular signatures associated with transformation and progression to breast cancer in the isogenic MCF10 model. Genomics 2008, 92, 419–428. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt Signaling in the Nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef]
- Yochum, G.S.; Cleland, R.; Goodman, R.H. A Genome-Wide Screen for β-Catenin Binding Sites Identifies a Downstream Enhancer Element That Controls c-Myc Gene Expression. Mol. Cell. Biol. 2008, 28, 7368–7379. [Google Scholar] [CrossRef] [PubMed]
- Bottomly, D.; Kyler, S.L.; McWeeney, S.K.; Yochum, G.S. Identification of β-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010, 38, 5735–5745. [Google Scholar] [CrossRef] [PubMed]
- Labak, C.M.; Wang, P.Y.; Arora, R.; Guda, M.R.; Asuthkar, S.; Tsung, A.J.; Velpula, K.K. Glucose transport: Meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am. J. Cancer Res. 2016, 6, 1599–1608. [Google Scholar] [PubMed]
- Ancey, P.-B.; Contat, C.; Meylan, E. Glucose transporters in cancer: From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- De La Roche, M.; Ibrahim, A.E.K.; Mieszczanek, J.; Bienz, M. LEF1 and B9L shield β-catenin from inactivation by axin, desensitizing colorectal cancer cells to tankyrase inhibitors. Cancer Res. 2014, 74, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, F.C.; Klein, K.; Carson, B.B.; Katz, S.; Ekas, L.A.; Evans, S. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 5954–5963. [Google Scholar] [CrossRef] [PubMed]
- Li, V.S.W.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt Signaling through Inhibition of β-Catenin Degradation in an Intact Axin1 Complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Oshima, M.; Oshima, H.; Kitagawa, K.; Kobayashi, M.; Itakura, C.; Taketo, M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc. Natl. Acad. Sci. USA 1995, 92, 4482–4486. [Google Scholar] [CrossRef]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, K.; Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Onuma, K.; Ochiai, M.; Imai, T.; Hippo, Y. Shortcuts to intestinal carcinogenesis by genetic engineering in organoids. Cancer Sci. 2019, 110, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Toyama, K.; Shioya, H.; Ito, M.; Hirota, M.; Hasegawa, S.; Matsumoto, H.; Takano, H.; Akiyama, T.; Toyoshima, K.; et al. Rapid Colorectal Adenoma Formation Initiated by Conditional Targeting of the Apc Gene. Science 1997, 278, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Onuma, K.; Ochiai, M.; Orihashi, K.; Takahashi, M.; Imai, T.; Nakagama, H.; Hippo, Y. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc. Natl. Acad. Sci. USA 2013, 110, 11127–11132. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.; Vogelstein, B. Lessons from Hereditary Review Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Kobayashi, A.; Yamamoto, M.; Hayes, J.D. The Nrf3 transcription factor is a membrane-bound glycoprotein targeted to the endoplasmic reticulum through its N-terminal homology box 1 sequence. J. Biol. Chem. 2009, 284, 3195–3210. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Taniguchi, H.; Ito, Y.; Morita, T.; Karim, M.R.; Ohtake, N.; Fukagai, K.; Ito, T.; Okamuro, S.; Iemura, S.; et al. The CK2-Nrf1 axis controls the clearance of ubiquitinated proteins by regulating proteasome gene expression. Mol. Cell. Biol. 2013, 33, 3461–3472. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aono, S.; Hatanaka, A.; Hatanaka, A.; Gao, Y.; Hippo, Y.; Taketo, M.M.; Waku, T.; Kobayashi, A. β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3344. https://doi.org/10.3390/ijms20133344
Aono S, Hatanaka A, Hatanaka A, Gao Y, Hippo Y, Taketo MM, Waku T, Kobayashi A. β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells. International Journal of Molecular Sciences. 2019; 20(13):3344. https://doi.org/10.3390/ijms20133344
Chicago/Turabian StyleAono, Shiori, Ayari Hatanaka, Atsushi Hatanaka, Yue Gao, Yoshitaka Hippo, Makoto Mark Taketo, Tsuyoshi Waku, and Akira Kobayashi. 2019. "β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells" International Journal of Molecular Sciences 20, no. 13: 3344. https://doi.org/10.3390/ijms20133344
APA StyleAono, S., Hatanaka, A., Hatanaka, A., Gao, Y., Hippo, Y., Taketo, M. M., Waku, T., & Kobayashi, A. (2019). β-Catenin/TCF4 Complex-Mediated Induction of the NRF3 (NFE2L3) Gene in Cancer Cells. International Journal of Molecular Sciences, 20(13), 3344. https://doi.org/10.3390/ijms20133344