Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte


 , , ,
, , , 

Abstract

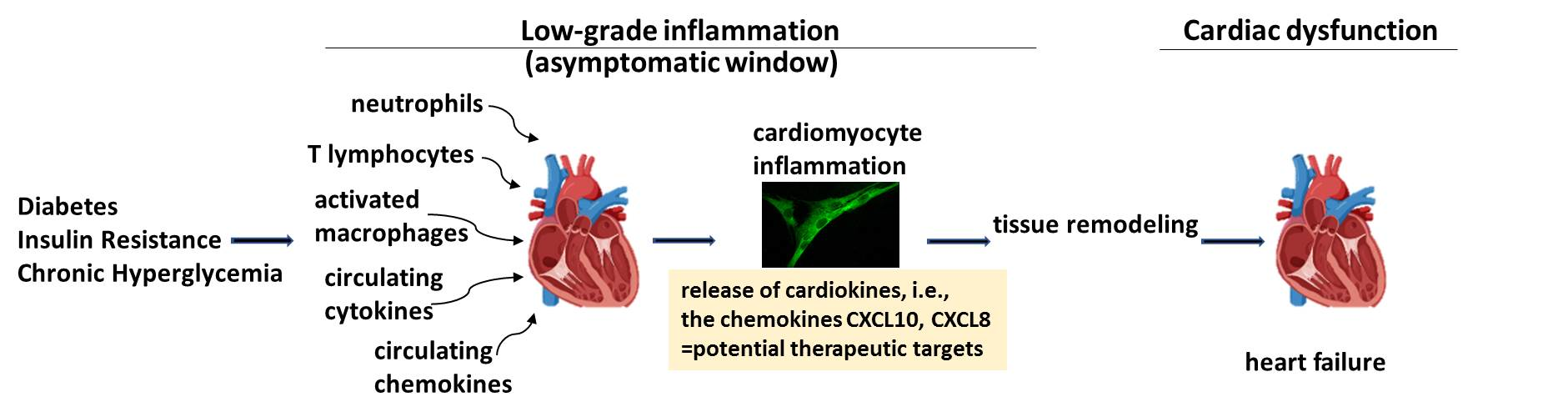{kind=link}
{kind=link}
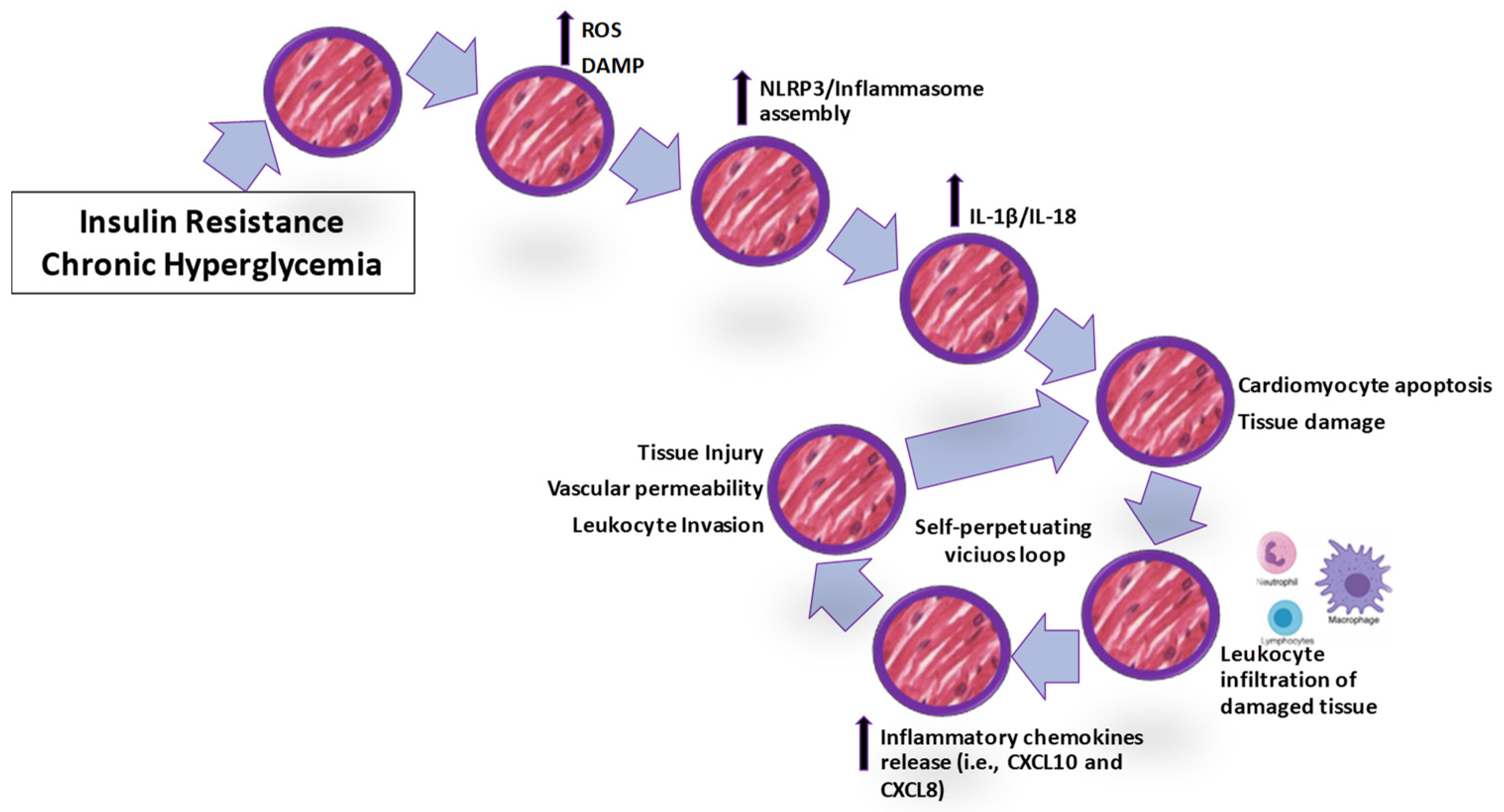{kind=link}
1. Introduction
2. DCM Etiology: The Pivotal Role of the Cardiomyocyte at Disease Onset
3. Cardiomyocyte Metabolism in Normal and Diabetic Condition
3.1. Metabolic Substrate Flexibility
3.2. Advanced Glycated End Products (AGE), Renin-Angiotensin-Aldosterone System (RAAS), Damage-Associated Molecular Pattern (DAMP) and Cardiomyocyte Damage
4. Cardiomyocyte Inflammation
4.1. The Inflammasome Platform
4.2. Leukocyte Infiltration in the Damaged Cardiomyocyte
5. The Cytokine Hypothesis
6. The Chemokines
CXCL10 and CXCL8 Potential Therapeutic Targets of PDE5i in DCM
7. An Overview of the Anti-Inflammatory Approach in DCM Treatment
8. A Window Opening on Sex-Dependent Molecular Mechanisms in the Cardiomyocyte Developing DCM
9. Conclusions
Funding
Conflicts of Interest
References
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Mizamtsidi, M.; Paschou, S.A.; Grapsa, J.; Vryonidou, A. Diabetic cardiomyopathy: A clinical entity or a cluster of molecular heart changes? Eur. J. Clin. Investig. 2016, 46, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Bernardo, B.C.; McMullen, J.R.; Ritchie, R.H. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol. Ther. 2014, 142, 375–415. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S. Diabetic cardiomyopathy: An expression of stage B heart failure with preserved ejection fraction. Diab. Vasc. Dis. Res. 2015, 12, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Castellano, J.M.; Farkouh, M.E.; Fuster, V. The relationships between cardiovascular disease and diabetes: Focus on pathogenesis. Endocrinol. Metab Clin. N. Am. 2014, 43, 41–57. [Google Scholar] [CrossRef]
- Poornima, I.G.; Parikh, P.; Shannon, R.P. Diabetic cardiomyopathy: The search for a unifying hypothesis. Circ. Res. 2006, 98, 596–605. [Google Scholar] [CrossRef]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia-and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef]
- von Lewinski, D.; Kolesnik, E.; Wallner, M.; Resl, M.; Sourij, H. New Antihyperglycemic Drugs and Heart Failure: Synopsis of Basic and Clinical Data. BioMed Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Borghetti, G.; von Lewinski, D.; Eaton, D.M.; Sourij, H.; Houser, S.R.; Wallner, M. Diabetic Cardiomyopathy: Current and Future Therapies. Beyond Glycemic Control. Front. Physiol. 2018, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Diamant, M.; Lamb, H.J.; Groeneveld, Y.; Endert, E.L.; Smit, J.W.; Bax, J.J.; Romijn, J.A.; de Roos, A.; Radder, J.K. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2003, 42, 328–335. [Google Scholar] [CrossRef]
- Wan, A.; Rodrigues, B. Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc. Res. 2016, 111, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Falcao-Pires, I.; Leite-Moreira, A.F. Diabetic cardiomyopathy: Understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail. Rev. 2012, 17, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; McNulty, P.; Taegtmeyer, H. Adaptation and maladaptation of the heart in diabetes: Part II: Potential mechanisms. Circulation 2002, 105, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Buttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef]
- Goyal, B.R.; Mehta, A.A. Diabetic cardiomyopathy: Pathophysiological mechanisms and cardiac dysfuntion. Hum. Exp. Toxicol. 2013, 32, 571–590. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Yin, R.; Caplan, M.J.; Hu, X.; Ren, J.; Shulman, G.I.; Sinusas, A.J.; Young, L.H. Additive effects of hyperinsulinemia and ischemia on myocardial GLUT1 and GLUT4 translocation in vivo. Circulation 1998, 98, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A.; Varela-Carver, A.; Mongillo, M.; Kleinert, C.; Khan, M.T.; Leccisotti, L.; Strickland, N.; Matsui, T.; Das, S.; Rosenzweig, A.; et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Heart J. 2010, 31, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Harmancey, R.; Lam, T.N.; Lubrano, G.M.; Guthrie, P.H.; Vela, D.; Taegtmeyer, H. Insulin resistance improves metabolic and contractile efficiency in stressed rat heart. FASEB J. 2012, 26, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Levelt, E.; Gulsin, G.; Neubauer, S.; McCann, G.P. MECHANISMS IN ENDOCRINOLOGY: Diabetic cardiomyopathy: Pathophysiology and potential metabolic interventions state of the art review. Eur. J. Endocrinol. 2018, 178, R127–R139. [Google Scholar] [CrossRef] [PubMed]
- Mandavia, C.H.; Aroor, A.R.; Demarco, V.G.; Sowers, J.R. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci. 2013, 92, 601–608. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, D.; Ren, J.; Yang, J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Rider, O.J.; Cox, P.; Tyler, D.; Clarke, K.; Neubauer, S. Myocardial substrate metabolism in obesity. Int. J. Obes. 2013, 37, 972–979. [Google Scholar] [CrossRef]
- Rodrigues, B.; Cam, M.C.; McNeill, J.H. Metabolic disturbances in diabetic cardiomyopathy. Mol. Cell. Biochem. 1998, 180, 53–57. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef] [PubMed]
- Mellor, K.M.; Ritchie, R.H.; Davidoff, A.J.; Delbridge, L.M. Elevated dietary sugar and the heart: Experimental models and myocardial remodeling. Can. J. Physiol. Pharmacol. 2010, 88, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, L.M.; Benson, V.L.; Ritchie, R.H.; Mellor, K.M. Diabetic Cardiomyopathy: The Case for a Role of Fructose in Disease Etiology. Diabetes 2016, 65, 3521–3528. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, A.; Obata, T.; Suzaki, M.; Takagi, Y.; Kida, Y.; Ogawa, T.; Tanaka, Y.; Asahina, T.; Ikebuchi, M.; Saeki, Y.; et al. Increase in cardiac muscle fructose content in streptozotocin-induced diabetic rats. Metabolism 1992, 41, 1041–1046. [Google Scholar] [CrossRef]
- Lal, S.; Randall, W.C.; Taylor, A.H.; Kappler, F.; Walker, M.; Brown, T.R.; Szwergold, B.S. Fructose-3-phosphate production and polyol pathway metabolism in diabetic rat hearts. Metabolism 1997, 46, 1333–1338. [Google Scholar] [CrossRef]
- Kayama, Y.; Raaz, U.; Jagger, A.; Adam, M.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic Cardiovascular Disease Induced by Oxidative Stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Bertoni, A.G.; Goff, D.C., Jr.; D’Agostino, R.B., Jr.; Liu, K.; Hundley, W.G.; Lima, J.A.; Polak, J.F.; Saad, M.F.; Szklo, M.; Tracy, R.P.; et al. Diabetic cardiomyopathy and subclinical cardiovascular disease: The Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes Care 2006, 29, 588–594. [Google Scholar] [CrossRef]
- Izzicupo, P.; Ghinassi, B.; D’Amico, M.A.; Di Blasio, A.; Gesi, M.; Napolitano, G.; Gallina, S.; Di Baldassarre, A. Effects of ACE I/D polymorphism and aerobic training on the immune-endocrine network and cardiovascular parameters of postmenopausal women. J. Clin. Endocrinol. Metab. 2013, 98, 4187–4194. [Google Scholar] [CrossRef]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Evans, T.; Mukherjee, K.; Karmazyn, M.; Chakrabarti, S. Diabetes-induced myocardial structural changes: Role of endothelin-1 and its receptors. J. Mol. Cell. Cardiol. 2000, 32, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial cell death in human diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Wold, L.E.; Sowers, J.R.; Quinn, M.T.; Ren, J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: Role of the AT1 receptor and NADPH oxidase. Hypertension 2003, 42, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Izzicupo, P.; Di Valerio, V.; MA, D.A.; Di Mauro, M.; Pennelli, A.; Falone, S.; Alberti, G.; Amicarelli, F.; Miscia, S.; Gallina, S.; et al. NAD(P)H oxidase and pro-inflammatory response during maximal exercise: Role of C242T polymorphism of the P22PHOX subunit. Int. J. Immunopathol. Pharmacol. 2010, 23, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef]
- Frieler, R.A.; Mortensen, R.M. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 2015, 131, 1019–1030. [Google Scholar] [CrossRef]
- Izzicupo, P.; D’Amico, M.A.; Bascelli, A.; Di Fonso, A.; D’Angelo, E.; Di Blasio, A.; Bucci, I.; Napolitano, G.; Gallina, S.; Di Baldassarre, A. Walking training affects dehydroepiandrosterone sulfate and inflammation independent of changes in spontaneous physical activity. Menopause 2013, 20, 455–463. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Corsini, E.; Galbiati, V.; Nikitovic, D.; Tsatsakis, A.M. Role of oxidative stress in chemical allergens induced skin cells activation. Food Chem. Toxicol. 2013, 61, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.C.; Chao, L.K.; Chou, J.C.; Dong, W.C.; Lin, C.N.; Lin, C.Y.; Chen, A.; Ka, S.M.; Ho, C.L.; Hua, K.F. Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1beta secretion. Inflamm. Res. 2013, 62, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Somanna, N.K.; Yariswamy, M.; Garagliano, J.M.; Siebenlist, U.; Mummidi, S.; Valente, A.J.; Chandrasekar, B. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell. Signal. 2015, 27, 1928–1938. [Google Scholar] [CrossRef]
- Santiago, J.J.; McNaughton, L.J.; Koleini, N.; Ma, X.; Bestvater, B.; Nickel, B.E.; Fandrich, R.R.; Wigle, J.T.; Freed, D.H.; Arora, R.C.; et al. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS ONE 2014, 9, e97281. [Google Scholar] [CrossRef] [PubMed]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Kuethe, F.; Sigusch, H.H.; Bornstein, S.R.; Hilbig, K.; Kamvissi, V.; Figulla, H.R. Apoptosis in patients with dilated cardiomyopathy and diabetes: A feature of diabetic cardiomyopathy? Horm. Metab. Res. 2007, 39, 672–676. [Google Scholar] [CrossRef]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femmino, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid. Med. Cell. Longev. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, Y.; Zhang, M.; An, F. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc. Drugs Ther. 2014, 28, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Sweet NETs, Bitter Wounds. Immunity 2015, 43, 223–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Silk, E.; Zhao, H.; Weng, H.; Ma, D. The role of extracellular histone in organ injury. Cell Death Dis. 2017, 8, e2812. [Google Scholar] [CrossRef]
- DeBerge, M.; Zhang, S.; Glinton, K.; Grigoryeva, L.; Hussein, I.; Vorovich, E.; Ho, K.; Luo, X.; Thorp, E.B. Efferocytosis and Outside-In Signaling by Cardiac Phagocytes. Links to Repair, Cellular Programming, and Intercellular Crosstalk in Heart. Front. Immunol. 2017, 8, 1428. [Google Scholar] [CrossRef]
- Khanna, S.; Biswas, S.; Shang, Y.; Collard, E.; Azad, A.; Kauh, C.; Bhasker, V.; Gordillo, G.M.; Sen, C.K.; Roy, S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE 2010, 5, e9539. [Google Scholar] [CrossRef]
- Tan, J.S.; Anderson, J.L.; Watanakunakorn, C.; Phair, J.P. Neutrophil dysfunction in diabetes mellitus. J. Lab. Clin. Med. 1975, 85, 26–33. [Google Scholar]
- Jakelic, J.; Kokic, S.; Hozo, I.; Maras, J.; Fabijanic, D. Nonspecific immunity in diabetes: Hyperglycemia decreases phagocytic activity of leukocytes in diabetic patients. Med. Arh. 1995, 49, 9–12. [Google Scholar]
- Rao, X.; Zhong, J.; Sun, Q. The heterogenic properties of monocytes/macrophages and neutrophils in inflammatory response in diabetes. Life Sci. 2014, 116, 59–66. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velazquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ. Heart Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.X.; Li, W.J.; Lu, Y.R.; Qin, J.; Wu, C.L.; Tian, M.; He, T.Y.; Yi, S.N.; Tang, D.Q.; Sun, L.; et al. Increased peripheral proinflammatory T helper subsets contribute to cardiovascular complications in diabetic patients. Mediat. Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef]
- Guzman-Flores, J.M.; Portales-Perez, D.P. Mechanisms of suppression of regulatory T-cells (Treg). Gac. Med. Mex. 2013, 149, 630–638. [Google Scholar]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Seta, Y.; Shan, K.; Bozkurt, B.; Oral, H.; Mann, D.L. Basic mechanisms in heart failure: The cytokine hypothesis. J. Card. Fail. 1996, 2, 243–249. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Durand, J.B.; Bies, R.D.; Young, J.B.; Mann, D.L. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation 1996, 93, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, J.; Mann, D.L. Positioning of inflammatory biomarkers in the heart failure landscape. J. Cardiovasc. Transl. Res. 2013, 6, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, A.; Tilley, D.G. The Role of Leukocytes in Diabetic Cardiomyopathy. Front. Physiol. 2018, 9, 1547. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Cavalera, M.; Wang, J.; Russo, I.; Shinde, A.; Kong, P.; Gonzalez-Quesada, C.; Rai, V.; Dobaczewski, M.; Lee, D.W.; et al. Smad3 Signaling Promotes Fibrosis While Preserving Cardiac and Aortic Geometry in Obese Diabetic Mice. Circ. Heart Fail. 2015, 8, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Dinh, W.; Futh, R.; Nickl, W.; Krahn, T.; Ellinghaus, P.; Scheffold, T.; Bansemir, L.; Bufe, A.; Barroso, M.C.; Lankisch, M. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc. Diabetol. 2009, 8, 58. [Google Scholar] [CrossRef]
- Masters, S.L.; Latz, E.; O’Neill, L.A. The inflammasome in atherosclerosis and type 2 diabetes. Sci. Transl. Med. 2011, 3, 81ps17. [Google Scholar] [CrossRef]
- Sivasubramanian, N.; Coker, M.L.; Kurrelmeyer, K.M.; MacLellan, W.R.; DeMayo, F.J.; Spinale, F.G.; Mann, D.L. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 2001, 104, 826–831. [Google Scholar] [CrossRef]
- Hokama, J.Y.; Ritter, L.S.; Davis-Gorman, G.; Cimetta, A.D.; Copeland, J.G.; McDonagh, P.F. Diabetes enhances leukocyte accumulation in the coronary microcirculation early in reperfusion following ischemia. J. Diabetes Complicat. 2000, 14, 96–107. [Google Scholar] [CrossRef]
- Pettersson, U.S.; Christoffersson, G.; Massena, S.; Ahl, D.; Jansson, L.; Henriksnas, J.; Phillipson, M. Increased recruitment but impaired function of leukocytes during inflammation in mouse models of type 1 and type 2 diabetes. PLoS ONE 2011, 6, e22480. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.P.; Morrow, V.A.; Brandie, F.M.; Connell, J.M.; Petrie, J.R. High glucose inhibits insulin-stimulated nitric oxide production without reducing endothelial nitric-oxide synthase Ser1177 phosphorylation in human aortic endothelial cells. J. Biol. Chem. 2003, 278, 18791–18797. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S.; Sullivan, L.M.; Roubenoff, R.; Dinarello, C.A.; Harris, T.; Benjamin, E.J.; Sawyer, D.B.; Levy, D.; Wilson, P.W.; D’Agostino, R.B.; et al. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: The Framingham Heart Study. Circulation 2003, 107, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Testa, M.; Yeh, M.; Lee, P.; Fanelli, R.; Loperfido, F.; Berman, J.W.; LeJemtel, T.H. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol. 1996, 28, 964–971. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef]
- Doroudgar, S.; Glembotski, C.C. The cardiokine story unfolds: Ischemic stress-induced protein secretion in the heart. Trends Mol. Med. 2011, 17, 207–214. [Google Scholar] [CrossRef]
- Tsutamoto, T.; Wada, A.; Ohnishi, M.; Tsutsui, T.; Ishii, C.; Ohno, K.; Fujii, M.; Matsumoto, T.; Yamamoto, T.; Takayama, T.; et al. Transcardiac increase in tumor necrosis factor-alpha and left ventricular end-diastolic volume in patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2004, 6, 173–180. [Google Scholar] [CrossRef]
- Nomiyama, H.; Osada, N.; Yoshie, O. The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 2010, 21, 253–262. [Google Scholar] [CrossRef]
- Romagnani, P.; Crescioli, C. CXCL10: A candidate biomarker in transplantation. Clin. Chim. Acta 2012, 413, 1364–1373. [Google Scholar] [CrossRef]
- Dusi, V.; Ghidoni, A.; Ravera, A.; De Ferrari, G.M.; Calvillo, L. Chemokines and Heart Disease: A Network Connecting Cardiovascular Biology to Immune and Autonomic Nervous Systems. Mediat. Inflamm. 2016, 2016, 5902947. [Google Scholar] [CrossRef]
- Di Luigi, L.; Corinaldesi, C.; Colletti, M.; Scolletta, S.; Antinozzi, C.; Vannelli, G.B.; Giannetta, E.; Gianfrilli, D.; Isidori, A.M.; Migliaccio, S.; et al. Phosphodiesterase Type 5 Inhibitor Sildenafil Decreases the Proinflammatory Chemokine CXCL10 in Human Cardiomyocytes and in Subjects with Diabetic Cardiomyopathy. Inflammation 2016, 39, 1238–1252. [Google Scholar] [CrossRef]
- Giannattasio, S.; Corinaldesi, C.; Colletti, M.; Di Luigi, L.; Antinozzi, C.; Filardi, T.; Scolletta, S.; Basili, S.; Lenzi, A.; Morano, S.; et al. The phosphodiesterase 5 inhibitor sildenafil decreases the proinflammatory chemokine IL-8 in diabetic cardiomyopathy: In vivo and in vitro evidence. J. Endocrinol. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Polverini, P.J.; Arenberg, D.A.; Kunkel, S.L. The role of CXC chemokines as regulators of angiogenesis. Shock 1995, 4, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; von Andrian, U.H. Chemokines in innate and adaptive host defense: Basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 2004, 22, 891–928. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. E. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, M.; Gerber, B.; Loetscher, P.; Jones, S.A.; Piali, L.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. Chemokine receptor specific for IP10 and mig: Structure, function, and expression in activated T-lymphocytes. J. Exp. Med. 1996, 184, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Luster, A.D.; Greenberg, S.M.; Leder, P. The IP-10 chemokine binds to a specific cell surface heparan sulfate site shared with platelet factor 4 and inhibits endothelial cell proliferation. J. Exp. Med. 1995, 182, 219–231. [Google Scholar] [CrossRef]
- Ohmori, Y.; Hamilton, T.A. Cell type and stimulus specific regulation of chemokine gene expression. Biochem. Biophys. Res. Commun. 1994, 198, 590–596. [Google Scholar] [CrossRef]
- Ohmori, Y.; Hamilton, T.A. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J. Immunol. 1995, 154, 5235–5244. [Google Scholar]
- van den Borne, P.; Quax, P.H.; Hoefer, I.E.; Pasterkamp, G. The multifaceted functions of CXCL10 in cardiovascular disease. BioMed Res. Int. 2014, 2014, 893106. [Google Scholar] [CrossRef]
- Hancock, W.W.; Lu, B.; Gao, W.; Csizmadia, V.; Faia, K.; King, J.A.; Smiley, S.T.; Ling, M.; Gerard, N.P.; Gerard, C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 2000, 192, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Hancock, W.W.; Gao, W.; Csizmadia, V.; Faia, K.L.; Shemmeri, N.; Luster, A.D. Donor-derived IP-10 initiates development of acute allograft rejection. J. Exp. Med. 2001, 193, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Szentes, V.; Gazdag, M.; Szokodi, I.; Dezsi, C.A. The Role of CXCR3 and Associated Chemokines in the Development of Atherosclerosis and During Myocardial Infarction. Front. Immunol. 2018, 9, 1932. [Google Scholar] [CrossRef] [PubMed]
- Scolletta, S.; Buonamano, A.; Sottili, M.; Giomarelli, P.; Biagioli, B.; Vannelli, G.B.; Serio, M.; Romagnani, P.; Crescioli, C. CXCL10 release in cardiopulmonary bypass: An in vivo and in vitro study. BioMed Aging Pathol. 2012, 2, 187–194. [Google Scholar] [CrossRef]
- Altara, R.; Manca, M.; Hessel, M.H.; Gu, Y.; van Vark, L.C.; Akkerhuis, K.M.; Staessen, J.A.; Struijker-Boudier, H.A.; Booz, G.W.; Blankesteijn, W.M. CXCL10 Is a Circulating Inflammatory Marker in Patients with Advanced Heart Failure: A Pilot Study. J. Cardiovasc. Transl. Res. 2016, 9, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Altara, R.; Mallat, Z.; Booz, G.W.; Zouein, F.A. The CXCL10/CXCR3 Axis and Cardiac Inflammation: Implications for Immunotherapy to Treat Infectious and Noninfectious Diseases of the Heart. J. Immunol. Res. 2016, 2016, 4396368. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Cassatella, M.A. Neutrophil-Derived Cytokines Involved in Physiological and Pathological Angiogenesis. Chemical Immunology Allergy; Karger Publishers: Basel, Switzerland, 2014; Volume 99, pp. 123–137. [Google Scholar] [CrossRef]
- Apostolakis, S.; Vogiatzi, K.; Amanatidou, V.; Spandidos, D.A. Interleukin 8 and cardiovascular disease. Cardiovasc. Res. 2009, 84, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Madan, A.K.; Pedersen, S.B.; Schiott, K.M.; Fain, J.N.; Richelsen, B. Higher production of IL-8 in visceral vs. subcutaneous adipose tissue. Implication of nonadipose cells in adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E8–E13. [Google Scholar] [CrossRef]
- Velasquez, I.M.; Frumento, P.; Johansson, K.; Berglund, A.; de Faire, U.; Leander, K.; Gigante, B. Association of interleukin 8 with myocardial infarction: Results from the Stockholm Heart Epidemiology Program. Int. J. Cardiol. 2014, 172, 173–178. [Google Scholar] [CrossRef]
- Cimini, F.A.; Barchetta, I.; Porzia, A.; Mainiero, F.; Costantino, C.; Bertoccini, L.; Ceccarelli, V.; Morini, S.; Baroni, M.G.; Lenzi, A.; et al. Circulating IL-8 levels are increased in patients with type 2 diabetes and associated with worse inflammatory and cardiometabolic profile. Acta Diabetol. 2017, 54, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Rothenbacher, D.; Muller-Scholze, S.; Herder, C.; Koenig, W.; Kolb, H. Differential expression of chemokines, risk of stable coronary heart disease, and correlation with established cardiovascular risk markers. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, C. The role of immunological biomarkers in cardiac rejection. Curr. Opin. Organ. Transplant. 2013, 18, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, C. Chemokines and transplant outcome. Clin. Biochem. 2016, 49, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, C.; Buonamano, A.; Scolletta, S.; Sottili, M.; Francalanci, M.; Giomarelli, P.; Biagioli, B.; Lisi, G.; Pradella, F.; Serio, M.; et al. Predictive role of pretransplant serum CXCL10 for cardiac acute rejection. Transplantation 2009, 87, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Kraaijeveld, A.O.; de Jager, S.C.; de Jager, W.J.; Prakken, B.J.; McColl, S.R.; Haspels, I.; Putter, H.; van Berkel, T.J.; Nagelkerken, L.; Jukema, J.W.; et al. CC chemokine ligand-5 (CCL5/RANTES) and CC chemokine ligand-18 (CCL18/PARC) are specific markers of refractory unstable angina pectoris and are transiently raised during severe ischemic symptoms. Circulation 2007, 116, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- de Jager, S.C.; Bongaerts, B.W.; Weber, M.; Kraaijeveld, A.O.; Rousch, M.; Dimmeler, S.; van Dieijen-Visser, M.P.; Cleutjens, K.B.; Nelemans, P.J.; van Berkel, T.J.; et al. Chemokines CCL3/MIP1alpha, CCL5/RANTES and CCL18/PARC are independent risk predictors of short-term mortality in patients with acute coronary syndromes. PLoS ONE 2012, 7, e45804. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Tuttolomondo, A.; Santilli, F.; Basili, S.; Ferrante, E.; Di Raimondo, D.; Pinto, A.; Licata, G. CD40 ligand and MCP-1 as predictors of cardiovascular events in diabetic patients with stroke. J. Atheroscler. Thromb. 2009, 16, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Canoui-Poitrine, F.; Luc, G.; Mallat, Z.; Machez, E.; Bingham, A.; Ferrieres, J.; Ruidavets, J.B.; Montaye, M.; Yarnell, J.; Haas, B.; et al. Systemic chemokine levels, coronary heart disease, and ischemic stroke events: The PRIME study. Neurology 2011, 77, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Sala, R.; Frati, C.; Lagrasta, C.; Madeddu, D.; Falco, A.; Pollino, S.; Bresciani, L.; Miragoli, M.; et al. Parenchymal and Stromal Cells Contribute to Pro-Inflammatory Myocardial Environment at Early Stages of Diabetes: Protective Role of Resveratrol. Nutrients 2016, 8, 729. [Google Scholar] [CrossRef] [PubMed]
- Taube, D.; Xu, J.; Yang, X.P.; Undrovinas, A.; Peterson, E.; Harding, P. Fractalkine depresses cardiomyocyte contractility. PLoS ONE 2013, 8, e69832. [Google Scholar] [CrossRef]
- Mandosi, E.; Giannetta, E.; Filardi, T.; Lococo, M.; Bertolini, C.; Fallarino, M.; Gianfrilli, D.; Venneri, M.A.; Lenti, L.; Lenzi, A.; et al. Endothelial dysfunction markers as a therapeutic target for Sildenafil treatment and effects on metabolic control in type 2 diabetes. Expert Opin. Ther. Targets 2015, 19, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Sottili, M.; Cosmi, L.; Borgogni, E.; Sarchielli, E.; Maggi, L.; Francalanci, M.; Vannelli, G.B.; Ronconi, E.; Adorini, L.; Annunziato, F.; et al. Immunomodulatory effects of BXL-01-0029, a less hypercalcemic vitamin D analogue, in human cardiomyocytes and T cells. Exp. Cell Res. 2009, 315, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, C.; Squecco, R.; Cosmi, L.; Sottili, M.; Gelmini, S.; Borgogni, E.; Sarchielli, E.; Scolletta, S.; Francini, F.; Annunziato, F.; et al. Immunosuppression in cardiac graft rejection: A human in vitro model to study the potential use of new immunomodulatory drugs. Exp. Cell Res. 2008, 314, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C.; Di Luigi, L.; Lenzi, A.; Crescioli, C. Phosphodiesterase type 5 inhibitors: Back and forward from cardiac indications. J. Endocrinol. Investig. 2016, 39, 143–151. [Google Scholar] [CrossRef]
- Moore, A.R.; Willoughby, D.A. The role of cAMP regulation in controlling inflammation. Clin. Exp. Immunol. 1995, 101, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.; Shah, K.B.; Hess, M.L. Phosphodiesterase inhibitors, congestive heart failure, and sudden death: Time for re-evaluation. Congest. Heart Fail. 2012, 18, 229–233. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Metra, M.; Zaca, V.; Sabbah, H.N.; Cotter, G.; Gheorghiade, M.; Cas, L.D. Agents with inotropic properties for the management of acute heart failure syndromes. Traditional agents and beyond. Heart Fail. Rev. 2009, 14, 243–253. [Google Scholar] [CrossRef]
- Rao, Y.J.; Xi, L. Pivotal effects of phosphodiesterase inhibitors on myocyte contractility and viability in normal and ischemic hearts. Acta Pharmacol. Sin. 2009, 30, 1–24. [Google Scholar] [CrossRef]
- Liu, H.; Maurice, D.H. Expression of cyclic GMP-inhibited phosphodiesterases 3A and 3B (PDE3A and PDE3B) in rat tissues: Differential subcellular localization and regulated expression by cyclic AMP. Br. J. Pharmacol. 1998, 125, 1501–1510. [Google Scholar] [CrossRef]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer’s disease. Neurotherapeutics 2015, 12, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.G.; Hutchings, D.C.; Woodward, M.; Rahimi, K.; Rutter, M.K.; Kirby, M.; Hackett, G.; Trafford, A.W.; Heald, A.H. Phosphodiesterase type-5 inhibitor use in type 2 diabetes is associated with a reduction in all-cause mortality. Heart 2016, 102, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Duranti, G.; Ceci, R.; Sgro, P.; Sabatini, S.; Di Luigi, L. Influence of the PDE5 inhibitor tadalafil on redox status and antioxidant defense system in C2C12 skeletal muscle cells. Cell Stress Chaperones 2017, 22, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Eng. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H., Jr.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Eng. J. Med. 2008, 358, 2545–2559. [Google Scholar]
- Kaplan, A.; Abidi, E.; El-Yazbi, A.; Eid, A.; Booz, G.W.; Zouein, F.A. Direct cardiovascular impact of SGLT2 inhibitors: Mechanisms and effects. Heart Fail. Rev. 2018, 23, 419–437. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Eng. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Desai, M.; Shaw, W.; Law, G.; Walton, M.K.; Rosenthal, N.; et al. Optimizing the analysis strategy for the CANVAS Program: A prespecified plan for the integrated analyses of the CANVAS and CANVAS-R trials. Diabetes Obes. Metab. 2017, 19, 926–935. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Kuschma, M.C.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Direct Cardiac Actions of Sodium Glucose Cotransporter 2 Inhibitors Target Pathogenic Mechanisms Underlying Heart Failure in Diabetic Patients. Front. Physiol. 2018, 9, 1575. [Google Scholar] [CrossRef]
- Custodio, J.S., Jr.; Duraes, A.R.; Abreu, M.; Albuquerque Rocha, N.; Roever, L. SGLT2 inhibition and heart failure-current concepts. Heart Fail. Rev. 2018, 23, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Schmidt, A.; Noutsias, M.; Song, X.Y.; Spillmann, F.; Riad, A.; Schultheiss, H.P.; et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol. 2007, 102, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Thomas, G.D.; Giroir, B.; Bryant, D.; Bullock, M.C.; Chwialkowski, M.C.; Victor, R.G.; Peshock, R.M. Magnetic resonance imaging and invasive evaluation of development of heart failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1999, 99, 448–454. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klein, B.; Brailly, H. Cytokine-binding proteins: Stimulating antagonists. Immunol. Today 1995, 16, 216–220. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report 2017. Available online: https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf (accessed on 22 May 2019).
- Norhammar, A.; Schenck-Gustafsson, K. Type 2 diabetes and cardiovascular disease in women. Diabetologia 2013, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef]
- Lum-Naihe, K.; Toedebusch, R.; Mahmood, A.; Bajwa, J.; Carmack, T.; Kumar, S.A.; Ardhanari, S.; DeMarco, V.G.; Emter, C.A.; Pulakat, L. Cardiovascular disease progression in female Zucker Diabetic Fatty rats occurs via unique mechanisms compared to males. Sci. Rep. 2017, 7, 17823. [Google Scholar] [CrossRef]
- Zhao, J.; Imbrie, G.A.; Baur, W.E.; Iyer, L.K.; Aronovitz, M.J.; Kershaw, T.B.; Haselmann, G.M.; Lu, Q.; Karas, R.H. Estrogen receptor-mediated regulation of microRNA inhibits proliferation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 257–265. [Google Scholar] [CrossRef]
- Toedebusch, R.; Belenchia, A.; Pulakat, L. Diabetic Cardiomyopathy: Impact of Biological Sex on Disease Development and Molecular Signatures. Front. Physiol. 2018, 9, 453. [Google Scholar] [CrossRef] [PubMed]
- Florijn, B.W.; Bijkerk, R.; van der Veer, E.P.; van Zonneveld, A.J. Gender and cardiovascular disease: Are sex-biased microRNA networks a driving force behind heart failure with preserved ejection fraction in women? Cardiovasc. Res. 2018, 114, 210–225. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filardi, T.; Ghinassi, B.; Di Baldassarre, A.; Tanzilli, G.; Morano, S.; Lenzi, A.; Basili, S.; Crescioli, C. Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte. Int. J. Mol. Sci. 2019, 20, 3299. https://doi.org/10.3390/ijms20133299
Filardi T, Ghinassi B, Di Baldassarre A, Tanzilli G, Morano S, Lenzi A, Basili S, Crescioli C. Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte. International Journal of Molecular Sciences. 2019; 20(13):3299. https://doi.org/10.3390/ijms20133299
Chicago/Turabian StyleFilardi, Tiziana, Barbara Ghinassi, Angela Di Baldassarre, Gaetano Tanzilli, Susanna Morano, Andrea Lenzi, Stefania Basili, and Clara Crescioli. 2019. "Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte" International Journal of Molecular Sciences 20, no. 13: 3299. https://doi.org/10.3390/ijms20133299
APA StyleFilardi, T., Ghinassi, B., Di Baldassarre, A., Tanzilli, G., Morano, S., Lenzi, A., Basili, S., & Crescioli, C. (2019). Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte. International Journal of Molecular Sciences, 20(13), 3299. https://doi.org/10.3390/ijms20133299