The Dualistic Effect of COX-2-Mediated Signaling in Obesity and Insulin Resistance


Abstract
1. Introduction
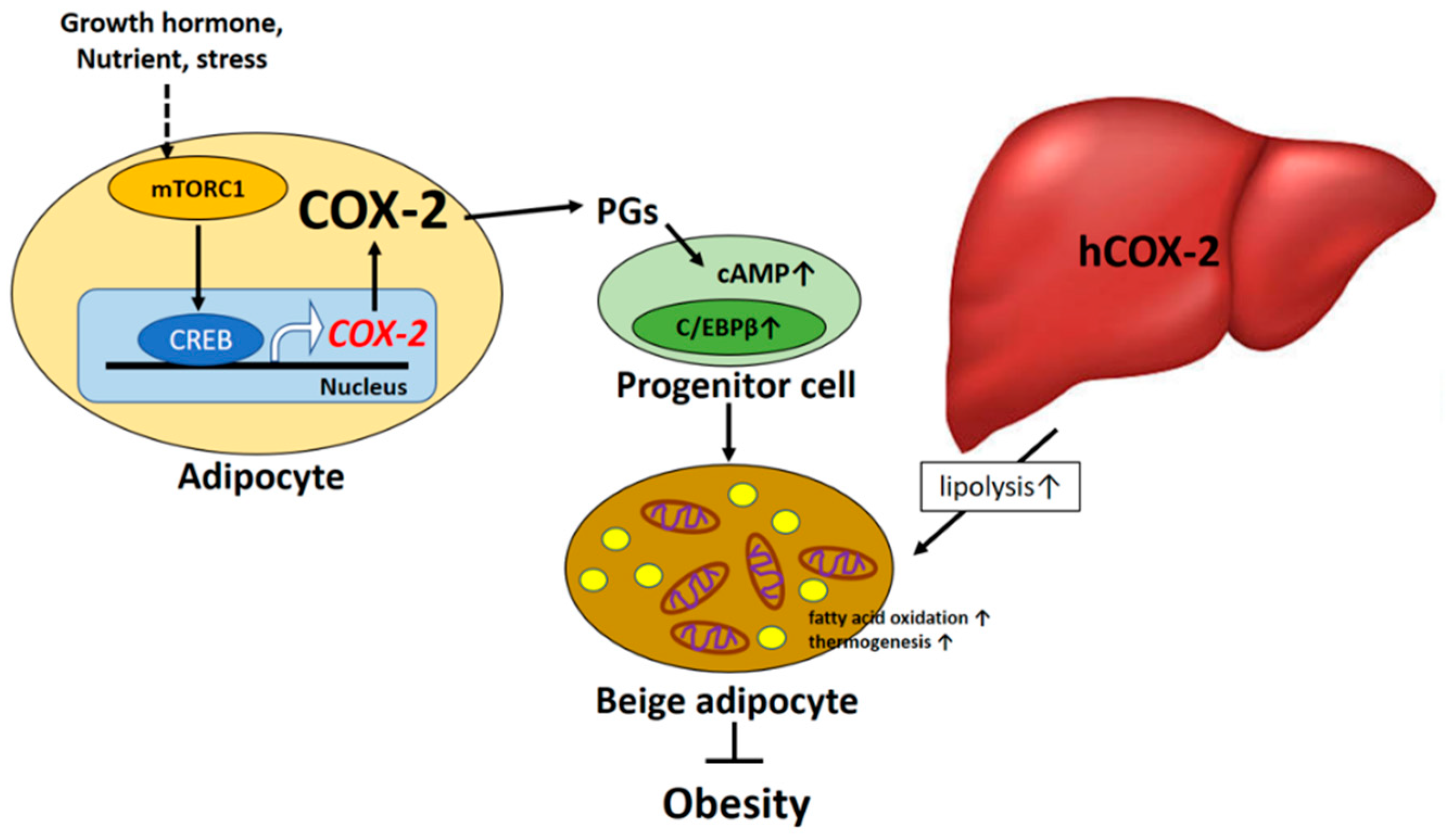
2. COX-2-Derived PGs and Regulation of Energy Metabolism
2.1. Adipose Tissue COX-2-Derived PGs and Regulation of Energy Metabolism
2.2. Hepatic COX-2-Derived PGs and Regulation of Energy Metabolism
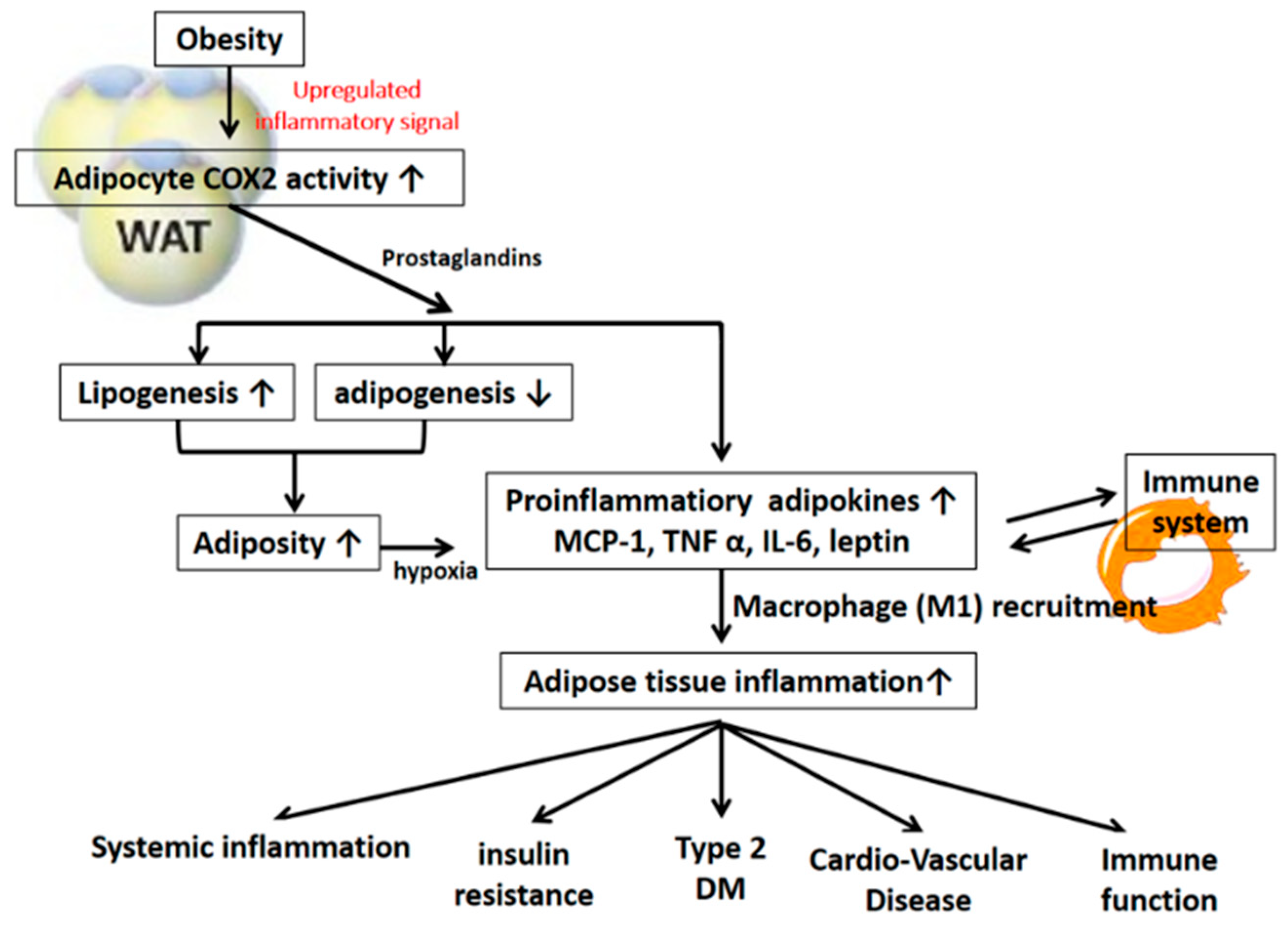
3. COX-2-Derived PGs and Obesity Associated Complications
3.1. COX-2-Derived PGs in the Development of Obesity and Adipogenesis
3.2. COX-2-Derived PGs in the Pathogenesis of Adipose Tissue Inflammation and Insulin Resistance
3.3. Hepatic COX-2-Derived PGs and Obesity and Insulin Resistance
3.4. COX-2-Derived PGs in Obesity Associated Cardiovascular Diseases
4. Clinical Implication
5. Conclusions and Future Direction
Funding
Conflicts of Interest
References
- Xu, H.; Fu, J.L.; Miao, Y.F.; Wang, C.J.; Han, Q.F.; Li, S.; Huang, S.Z.; Du, S.N.; Qiu, Y.X.; Yang, J.C.; et al. Prostaglandin E2 receptor EP3 regulates both adipogenesis and lipolysis in mouse white adipose tissue. J. Mol. Cell. Biol. 2016, 8, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta. 2015, 1851, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Madsen, L.; Pedersen, L.M.; Lillefosse, H.H.; Fjaere, E.; Bronstad, I.; Hao, Q.; Petersen, R.K.; Hallenborg, P.; Ma, T.; De Matteis, R.; et al. UCP1 induction during recruitment of brown adipocytes in white adipose tissue is dependent on cyclooxygenase activity. PLoS ONE 2010, 5, e11391. [Google Scholar] [CrossRef] [PubMed]
- Vegiopoulos, A.; Müller-Decker, K.; Strzoda, D.; Schmitt, I.; Chichelnitskiy, E.; Ostertag, A.; Berriel, D.M.; Rozman, J.; Hrabe de Angelis, M.; Nüsing, R.M.; et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010, 28, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Bayindir, I.; Babaeikelishomi, R.; Kocanova, S.; Sousa, I.S.; Lerch, S.; Hardt, O.; Wild, S.; Bosio, A.; Bystricky, K.; Herzig, S.; et al. Transcriptional Pathways in cPGI2-Induced Adipocyte Progenitor Activation for Browning. Front Endocrinol (Lausanne) 2015, 17, 129. [Google Scholar] [CrossRef]
- Ghandour, R.A.; Giroud, M.; Vegiopoulos, A.; Herzig, S.; Ailhaud, G.; Amri, E.Z.; Pisani, D.F. IP-receptor and PPARs trigger the conversion of human white to brite adipocyte induced by carbaprostacyclin. Biochim. Biophys. Acta 2016, 1861, 285–293. [Google Scholar] [CrossRef] [PubMed]
- García-Alonso, V.; López-Vicario, C.; Titos, E.; Morán-Salvador, E.; González-Périz, A.; Rius, B.; Párrizas, M.; Werz, O.; Arroyo, V.; Clària, J. Coordinate functional regulation between microsomal prostaglandin E synthase-1 (mPGES-1) and peroxisome proliferator-activated receptor γ (PPARγ) in the conversion of white-to-brown adipocytes. J. Biol. Chem. 2013, 288, 28230–28242. [Google Scholar] [CrossRef]
- García-Alonso, V.; Titos, E.; Alcaraz-Quiles, J.; Rius, B.; Lopategi, A.; López-Vicario, C.; Jakobsson, P.J.; Delgado, S.; Lozano, J.; Clària, J. Prostaglandin E2 Exerts Multiple Regulatory Actions on Human Obese Adipose Tissue Remodeling, Inflammation, Adaptive Thermogenesis and Lipolysis. PLoS ONE 2016, 11, e0153751. [Google Scholar]
- Zhang, X.; Luo, Y.; Wang, C.; Ding, X.; Yang, X.; Wu, D.; Silva, F.; Yang, Z.; Zhou, Q.; Wang, L.; et al. Adipose mTORC1 Suppresses Prostaglandin Signaling and Beige Adipogenesis via the CRTC2-COX-2 Pathway. Cell Rep. 2018, 24, 3180–3193. [Google Scholar] [CrossRef]
- Paschos, G.K.; Tang, S.Y.; Theken, K.N.; Li, X.; Verginadis, I.; Lekkas, D.; Herman, L.; Yan, W.; Lawson, J.; FitzGerald, G.A. Cold-Induced Browning of Inguinal White Adipose Tissue Is Independent of Adipose Tissue Cyclooxygenase-2. Cell Rep. 2018, 24, 809–814. [Google Scholar] [CrossRef]
- Francés, D.E.; Motiño, O.; Agrá, N.; González-Rodríguez, Á.; Fernández-Álvarez, A.; Cucarella, C.; Mayoral, R.; Castro-Sánchez, L.; García-Casarrubios, E.; Boscá, L.; et al. Hepatic cyclooxygenase-2 expression protects against diet-induced steatosis, obesity, and insulin resistance. Diabetes 2015, 64, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Konheim, Y.L.; Wolford, J.K. Association of a promoter variant in the inducible cyclooxygenase-2 gene (PTGS2) with type 2 diabetes mellitus in Pima Indians. Hum. Genet. 2003, 113, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Sánchez, A.; Pilar Martínez, M.; Benedito, S.; López-Oliva, M.E.; García-Sacristán, A.; Hernández, M.; Prieto, D. COX-2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic. Biol. Med. 2015, 84, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Kiritoshi, S.; Nishikawa, T.; Sonoda, K.; Kukidome, D.; Senokuchi, T.; Matsuo, T.; Matsumura, T.; Tokunaga, H.; Brownlee, M.; Araki, E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes 2003, 52, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Bowers, L.W.; de Graffenried, L.A. Targeting the COX-2 Pathway to Improve Therapeutic Response in the Obese Breast Cancer Patient Population. Curr. Pharmacol. Rep. 2015, 1, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Hsiao, F.C.; Chang, H.M.; Wabitsch, M.; Hsieh, P.S. Importance of adipocyte cyclooxygenase-2 and prostaglandin E2-prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 2016, 30, 2282–2297. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Leffler, C.W.; Cowan, G.S., Jr.; Buffington, C.; Pouncey, L.; Bahouth, S.W. Stimulation of leptin release by arachidonic acid and prostaglandin E(2) in adipose tissue from obese humans. Metabolism 2001, 50, 921–928. [Google Scholar] [CrossRef]
- Ghoshal, S.; Trivedi, D.B.; Graf, G.A.; Loftin, C.D. Cyclooxygenase-2 deficiency attenuates adipose tissue differentiation and inflammation in mice. J Biol Chem. 2011, 286, 889–898. [Google Scholar] [CrossRef]
- Fain, J.N. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm. 2006, 74, 443–477. [Google Scholar]
- Maihöfner, C.; Charalambous, M.P.; Bhambra, U.; Lightfoot, T.; Geisslinger, G.; Gooderham, N.J. Colorectal Cancer Group.; Expression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal cancer. Carcinogenesis 2003, 24, 665–671. [Google Scholar] [CrossRef]
- Jaworski, K.; Ahmadian, M.; Duncan, R.E.; Sarkadi-Nagy, E.; Varady, K.A.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; Kim, K.H.; et al. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med. 2009, 15, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Nishimura, K.; Jisaka, M.; Nagaya, T.; Shono, F.; Yokota, K. Up-regulation of adipogenesis in adipocytes expressing stably cyclooxygenase-2 in the antisense direction. Prostaglandins Other Lipid Mediat. 2010, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Miard, S.; Briggs, M.R.; Auwerx, J. Selective cyclo-oxygenase-2 inhibitors impair adipocyte differentiation through inhibition of the clonal expansion phase. J. Lipid Res. 2003, 44, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Inazumi, T.; Shirata, N.; Morimoto, K.; Takano, H.; Segi-Nishida, E.; Sugimoto, Y. Prostaglandin E₂-EP4 signaling suppresses adipocyte differentiation in mouse embryonic fibroblasts via an autocrine mechanism. J. Lipid Res. 2011, 52, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.S.; Jin, J.S.; Chiang, C.F.; Chan, P.C.; Chen, C.H.; Shih, K.C. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity (Silver Spring). 2009, 17, 1150–1157. [Google Scholar] [CrossRef]
- Hsieh, P.S.; Lu, K.C.; Chiang, C.F.; Chen, C.H. Suppressive effect of COX2 inhibitor on the progression of adipose inflammation in high-fat-induced obese rats. Eur. J. Clin. Invest. 2010, 40, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Cifarelli, V.; Sun, S.; Kuda, O.; Abumrad, N.A.; Su, X. Major role of adipocyte prostaglandin E2 in lipolysis-induced macrophage recruitment. J. Lipid Res. 2016, 57, 663–673. [Google Scholar] [CrossRef]
- Tian, Y.F.; Hsia, T.L.; Hsieh, C.H.; Huang, D.W.; Chen, C.H.; Hsieh, P.S. The importance of cyclooxygenase 2-mediated oxidative stress in obesity-induced muscular insulin resistance in high-fat-fed rats. Life Sci. 2011, 89, 107–114. [Google Scholar] [CrossRef]
- Granado, M.; Martín, A.I.; Castillero, E.; López-Calderón, A.; Villanúa, M.A. Cyclooxygenase-2 inhibition reverts the decrease in adiponectin levels and attenuates the loss of white adipose tissue during chronic inflammation. Eur. J. Pharmacol. 2009, 17, 97–103. [Google Scholar] [CrossRef]
- Lijnen, H.R.; Van Hoef, B.; Lu, H.R.; Gallacher, D.J. Rofecoxib impairs adipose tissue development in a murine model of nutritionally induced obesity. Thromb. Haemost. 2008, 100, 338–342. [Google Scholar]
- Pierre, C.; Guillebaud, F.; Airault, C.; Baril, N.; Barbouche, R.; Save, E.; Gaigé, S.; Bariohay, B.; Dallaporta, M.; Troadec, J.D. Invalidation of Microsomal Prostaglandin E Synthase-1 (mPGES-1) Reduces Diet-Induced Low-Grade Inflammation and Adiposity. Front. Physiol. 2018, 2, 1358. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.P.; Lee, D.; Maulis, M.F.; Carboneau, B.A.; Threadgill, D.W.; Poffenberger, G.; Milne, G.; Boyd, K.L.; Powers, A.C.; McGuinness, O.P.; et al. The PGE2 EP3 Receptor Regulates Diet-Induced Adiposity in Male Mice. Endocrinology 2016, 157, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, J.; Michaud, A.; Gauthier, M.F.; Pelletier, M.; Julien, F.; Bouvet-Bouchard, L.; Tchernof, A. Interleukin-1β and prostaglandin-synthesizing enzymes as modulators of human omental and subcutaneous adipose tissue function. Prostaglandins Leukot. Essent. Fatty Acids 2019, 141, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Cucak, H.; Grunnet, L.G.; Rosendahl, A. Accumulation of M1-like macrophages in type 2 diabetic islets is followed by a systemic shift in macrophage polarization. J. Leukoc. Biol. 2014, 95, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Carli, C.; Metz, C.N.; Al-Abed, Y.; Naccache, P.H.; Akoum, A. Up-regulation of cyclooxygenase-2 expression and prostaglandin E2 production in human endometriotic cells by macrophage migration inhibitory factor: Involvement of novel kinase signaling pathways. Endocrinology 2009, 150, 3128–3137. [Google Scholar] [CrossRef] [PubMed]
- Sampey, A.V.; Hall, P.H.; Mitchell, R.A.; Metz, C.N.; Morand, E.F. Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum. 2001, 44, 1273–1280. [Google Scholar] [CrossRef]
- Chan, P.C.; Wu, T.N.; Chen, Y.C.; Lu, C.H.; Wabitsch, M.; Tian, Y.F.; Hsieh, P.S. Targetted inhibition of CD74 attenuates adipose COX-2-MIF-mediated M1 macrophage polarization and retards obesity-related adipose tissue inflammation and insulin resistance. Clin. Sci. (Lond). 2018, 132, 1581–1596. [Google Scholar] [CrossRef]
- Coll, T.; Palomer, X.; Blanco-Vaca., F.; Escolà-Gil, J.C.; Sánchez, R.M.; Laguna, J.C.; Vázquez-Carrera, M. Cyclooxygenase 2 inhibition exacerbates palmitate-induced inflammation and insulin resistance in skeletal muscle cells. Endocrinology 2010, 151, 537–548. [Google Scholar] [CrossRef][Green Version]
- Tsujimoto, S.; Kishina, M.; Koda, M.; Yamamoto, Y.; Tanaka, K.; Harada, Y.; Yoshida, A.; Hisatome, I. Nimesulide, a cyclooxygenase-2 selective inhibitor, suppresses obesity-related non-alcoholic fatty liver disease and hepatic insulin resistance through the regulation of peroxisome proliferator-activated receptor γ. Int. J. Mol. Med. 2016, 38, 721–728. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Bai, Q.; Song, J.; Guan, J.; Gao, J.; Liu, B.; Ma, X.; Du, Y. Celecoxib attenuates liver steatosis and inflammation in non-alcoholic steatohepatitis induced by high-fat diet in rats. Mol. Med. Rep. 2011, 4, 811–816. [Google Scholar] [PubMed]
- Motiño, O.; Agra, N.; Brea Contreras, R.; Domínguez-Moreno, M.; García-Monzón, C.; Vargas-Castrillón, J.; Carnovale, C.E.; Boscá, L.; Casado, M.; Mayoral, R.; et al. Cyclooxygenase-2 expression in hepatocytes attenuates non-alcoholic steatohepatitis and liver fibrosis in mice. Biochim. Biophys. Acta 2016, 1862, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Motiño, O.; Francés, D.E.; Mayoral, R.; Castro-Sánchez, L.; Fernández-Velasco, M.; Boscá, L.; García-Monzón, C.; Brea, R.; Casado, M.; Agra, N.; et al. Regulation of MicroRNA 183 by Cyclooxygenase 2 in Liver Is DEAD-Box Helicase p68 (DDX5) Dependent: Role in Insulin Signaling. Mol. Cell. Biol. 2015, 35, 2554–2567. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yang, L.X.; Guo, R.; Shi, Y.; Hou, X.; Yang, Z.; Zhou, X.; Liu, H. Atorvastatin attenuates plaque vulnerability by downregulation of EMMPRIN expression via COX-2/PGE2 pathway. Exp. Ther. Med. 2017, 13, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Raval, M.; Frank, P.G.; Laury-Kleintop, L.; Yan, G.; Lanza-Jacoby, S. Celecoxib combined with atorvastatin prevents progression of atherosclerosis. J. Surg. Res. 2010, 163, e113–e122. [Google Scholar] [CrossRef] [PubMed]
- Baldan, A.; Ferronato, S.; Olivato, S.; Malerba, G.; Scuro, A.; Veraldi, G.F.; Gelati, M.; Ferrari, S.; Mariotto, S.; Pignatti, P.F.; et al. Cyclooxygenase 2, toll-like receptor 4 and interleukin 1β mRNA expression in atherosclerotic plaques of type 2 diabetic patients. Inflamm. Res. 2014, 63, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Grosser, T.; Yu, Y.; Fitzgerald, G.A. Emotion recollected in tranquility: Lessons learned from the COX-2 saga. Annu. Rev. Med. 2010, 61, 17–33. [Google Scholar] [CrossRef]
- González-Ortiz, M.; Pascoe-González, S.; Hernández-Salazar, E.; Kam-Ramos, A.M. Effect of celecoxib, a cyclooxygenase-2-specific inhibitor, on insulin sensitivity, C-reactive protein, homocysteine, and metabolic profile in overweight or obese subjects. Metab. Syndr. Relat. Disord. 2005, 3, 95–101. [Google Scholar] [CrossRef]
- Lu, C.H.; Hung, Y.J.; Hsieh, P.S. Additional effect of metformin and celecoxib against lipid dysregulation and adipose tissue inflammation in high-fat fed rats with insulin resistance and fatty liver. Eur. J. Pharmacol. 2016, 15, 60–67. [Google Scholar] [CrossRef]
- Halpern, B.; Mancini, M.C. Safety assessment of combination therapies in the treatment of obesity: Focus on naltrexone/bupropion extended release and phentermine-topiramate extended release. Expert Opin. Drug Saf. 2017, 16, 27–39. [Google Scholar] [CrossRef]
- Lu, C.H.; Chung, C.H.; Lee, C.H.; Hsieh, C.H.; Hung, Y.J.; Lin, F.H.; Tsao, C.H.; Hsieh, P.S.; Chien, W.C. Combination COX-2 inhibitor and metformin attenuate rate of joint replacement in osteoarthritis with diabetes: A nationwide, retrospective, matched-cohort study in Taiwan. PLoS ONE 2018, 13, e0191242. [Google Scholar] [CrossRef] [PubMed]
- Micallef, D.; Micallef, S.; Schembri-Wismayer, P.; Calleja-Agius, J. Novel applications of COX-2 inhibitors, metformin, and statins for the primary chemoprevention of breast cancer. J. Turk. Ger. Gynecol. Assoc. 2016, 17, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, T.; Savolainen, M.J.; Jääskeläinen, T.; Kesäniemi, Y.A. Effect of a synthetic prostaglandin E2 analogue, RS-86505-007, on plasma lipids and lipoproteins in patients with moderate hypercholesterolaemia: Efficacy and tolerance of treatment and response in different apolipoprotein polymorphism groups. Eur. J. Clin. Pharmacol. 1995, 48, 97–102. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Gene | Model | Effects | Reference | |
|---|---|---|---|---|---|
| Energy metabolism | global | Cox-2 | COX2 KO mice | Cold-induced expression of UCP1 in inguinal white adipocytes was repressed | [3] |
| (B6;129P2 Ptgs2 tm1Unc) | |||||
| global | Cox-2 | COX2 KO mice | BAT characteristics were diminishedin WAT of CL-treated COX-2–/– mice | [4] | |
| (B6;129P2 Ptgs2 tm1Unc) | |||||
| global | Cox-2 | K5COX2 mice (line 675+/+) | induced de novo BAT recruitment in WAT, increased systemic energy expenditure, and protected mice against HFD–induced obesity. | [4] | |
| (overexpressing the COX-2 gene under the control of the promoter for the keratin 5 gene) | |||||
| global | Cox-2 | COX2 KO mice | Deletion of COX-2 fails to suppress cold-induced browning and UCP-1 expression in AT | [10] | |
| (COX-2 flox/flox mice; Cre-ER,tamoxifen-inducible form of Cre-recombinase) | |||||
| adipose tissue | Cox-2 | adipocyte-specific COX-2 KO mice | no alteration in metabolite excretion under basal conditions and augment their formation in response to cold | [10] | |
| (COX-2 flox/flox mice; adiponectin-Cre) | |||||
| Obesity and insulin resistance | global | AdPLA | AdPLA-null mice | increases lipolysis and prevents obesity induced by HFD feeding | [21] |
| (C57BL/ 6J) | |||||
| global | Cox-2 | COX-2−/− mice | Macrophage-dependent AT inflammation was reduced | [18] | |
| (C57BL/6J × 129/Ola (C57/129)) | |||||
| global | mPGES-1 | mPGES-1−/− (KO) mice | reduces diet-induced low-grade inflammation and adiposity | [31] | |
| (DBA/11ac J) | |||||
| global | EP3 | EP3−/− mice | increased epididymal fat mass and adipocyte size and macrophage infiltration | [32] | |
| (C57BL/6J) | |||||
| liver | Cox-2 | hepatocyte-specific COX-2 transgenic mice | lower grades of steatosis, inflammation and reduced recruitment and infiltration of hepatic macrophages | [43] | |
| (B6D2/OlaHsd) | |||||
| global | Cox-2/PGE2/EP3 | HFD induced obese mice | increased obesity-associated AT inflammation and systemic insulin resistance | [16,25,26] |
| Target Gene or Protein | Method | Model | Effects | Reference | |
|---|---|---|---|---|---|
| Energy metabolism | COX | COX inhibitor, indomethacin | Rb–/– MEFs | COX activity is required for induction of UCP-1 | [3] |
| (embryo fibroblasts (MEFs) lacking the retinoblastioma (Rb) gene) | |||||
| cPGI2 | norepinephrine (NE) treatment | Differentiation of primary human mesenchymal cells | NE-induced cPGI2 shifts the differentiation of WAT mesenchymal progenitors toward a brown adipocyte phenotype | [4,5] | |
| cPGI2 | norepinephrine (NE) treatment | beige/brite progenitor cells | cPGI2 induces a broad thermogenic gene expression program in adipocyte progenitors | [5] | |
| (Lin−CD29+CD34+Sca-1+ cells) | |||||
| cPGI2 | cPGI2 treatment | hMADS | activates white to brite adipocyte conversion | [6] | |
| (human multipotent adipose-derived stem cells) | |||||
| mPGES-1 | mPGES-1 siRNA | 3T3-L1 adipocytes | mPGES-1 as a key regulator of white-to-brown adipogenesis | [7] | |
| PGE2 | PGE2 treatment | adipocytes isolated from human omental WAT | PGE2 increased the expression of UCP1 and PRDM16 in adipocytes | [8] | |
| Obesity and insulin resistance | Cox-2 | COX-2 shRNA and COX-2 inhibitor, NS398 | 3T3-L1 adipocytes | The suppressive effect of COX-2 inhibition was noted in the release of pro-inflammatory adipokines into the medium from the hypertrophy adipocytes | [16] |
| Cox-2 | lentivirus derived shCOX-2 or COX-2 cDNA | SGBS adipocytes | adipocyte COX-2 activation up-regulates MIF production during thedevelopment of hypertrophy and hypoxia | [38] | |
| Cox-2 | COX-2 inhibitor,sc-58236 | 3T3-L1 adipocytes | inhibition of the COX-2 enzyme impairs adipocyte differentiation | [23] | |
| Cox-2 | COX-2 inhibitor, NS398 | mouse embryonic fibroblasts (MEF) | COX-2-derived PGE2 suppresses adipocyte differentiation in MEF cells | [24] | |
| EP3 | mouse embryonic fibroblasts (MEF) isolated EP3–/– mice WAT | activation of EP3 receptor suppressed adipogenesis and lipolysis | [1] |
| 1 | The cellular and molecular mechanisms and the potential interplay of COX-2 derived PGs in control of energy metabolism and the development of obesity and insulin resistance. |
| 2 | The detailed mechanisms regarding the role of PGs and their receptors in the development of these COX-2 mediated phenomenon. |
| 3 | The therapeutive strategy to develop COX-2 targeting compounds which could boost energy expenditure without trigger COX-2-mediated inflammation |
| 4 | To dissect the role of COX-2 dependent and independent adaptive thermogenesis and their impact on energy homeostasis |
| 5 | Clinical application of selective COX-2 activator in prevention and treatment of NAFLD |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, P.-C.; Liao, M.-T.; Hsieh, P.-S. The Dualistic Effect of COX-2-Mediated Signaling in Obesity and Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 3115. https://doi.org/10.3390/ijms20133115
Chan P-C, Liao M-T, Hsieh P-S. The Dualistic Effect of COX-2-Mediated Signaling in Obesity and Insulin Resistance. International Journal of Molecular Sciences. 2019; 20(13):3115. https://doi.org/10.3390/ijms20133115
Chicago/Turabian StyleChan, Pei-Chi, Min-Tser Liao, and Po-Shiuan Hsieh. 2019. "The Dualistic Effect of COX-2-Mediated Signaling in Obesity and Insulin Resistance" International Journal of Molecular Sciences 20, no. 13: 3115. https://doi.org/10.3390/ijms20133115
APA StyleChan, P.-C., Liao, M.-T., & Hsieh, P.-S. (2019). The Dualistic Effect of COX-2-Mediated Signaling in Obesity and Insulin Resistance. International Journal of Molecular Sciences, 20(13), 3115. https://doi.org/10.3390/ijms20133115