Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis


Abstract
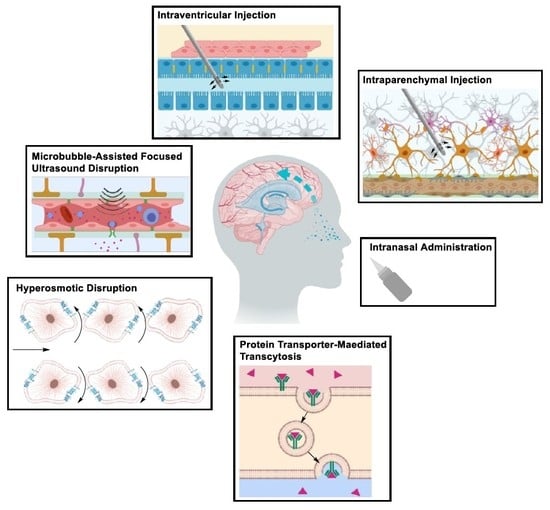
1. Introduction
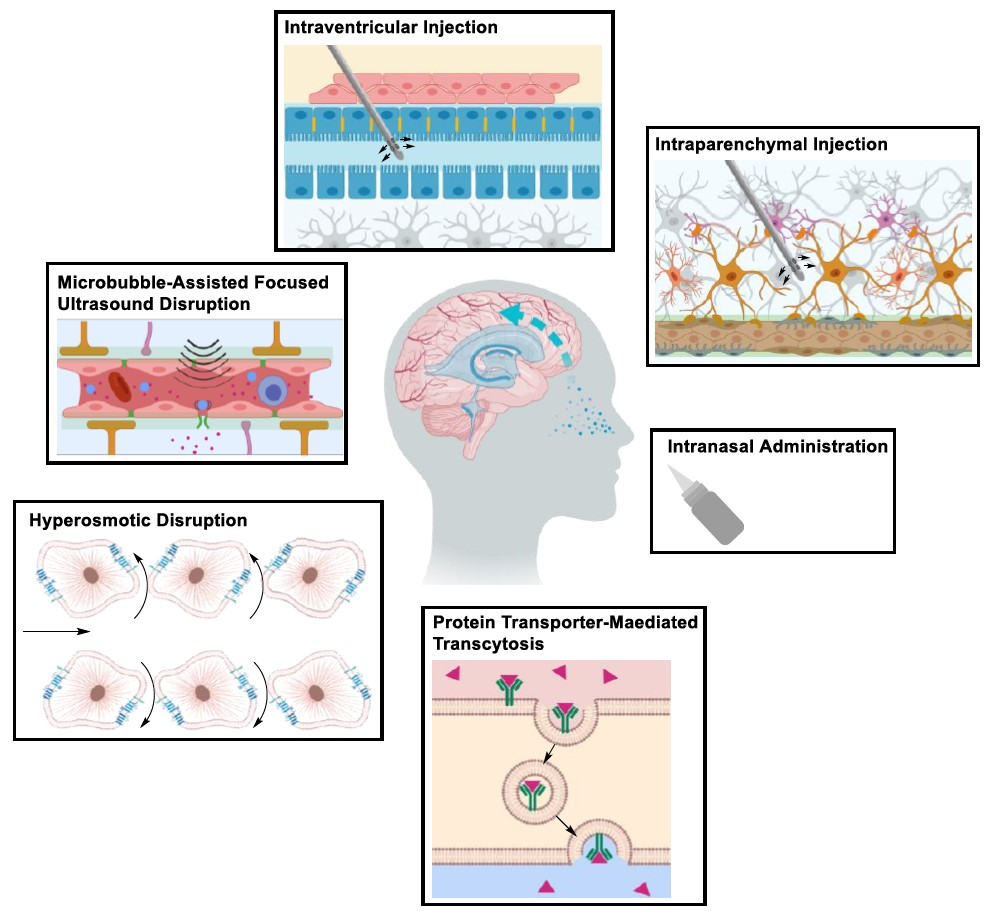
2. Structural Components of the BBB
2.1. Strategies for Delivery of Therapeutics Across the BBB
2.1.1. Drug Delivery Beyond the BBB
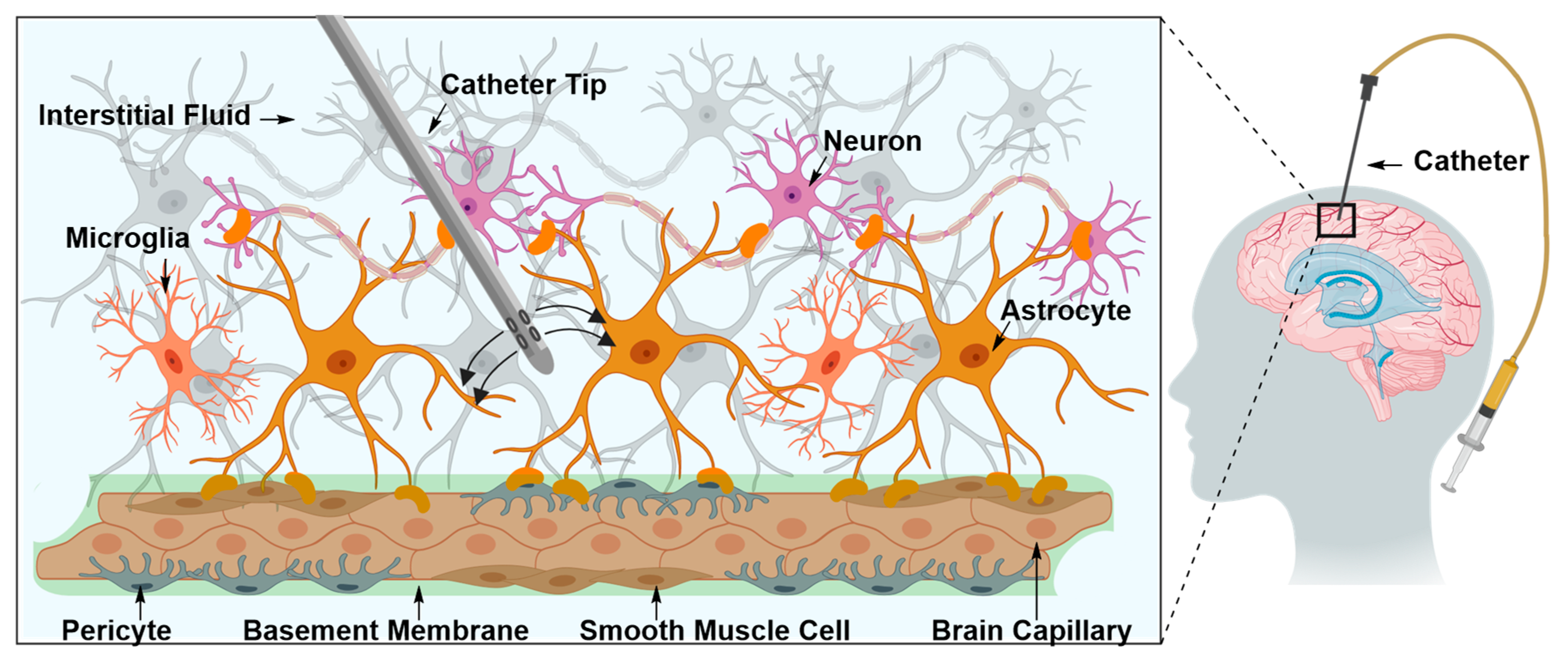
Intraparenchymal Drug Delivery
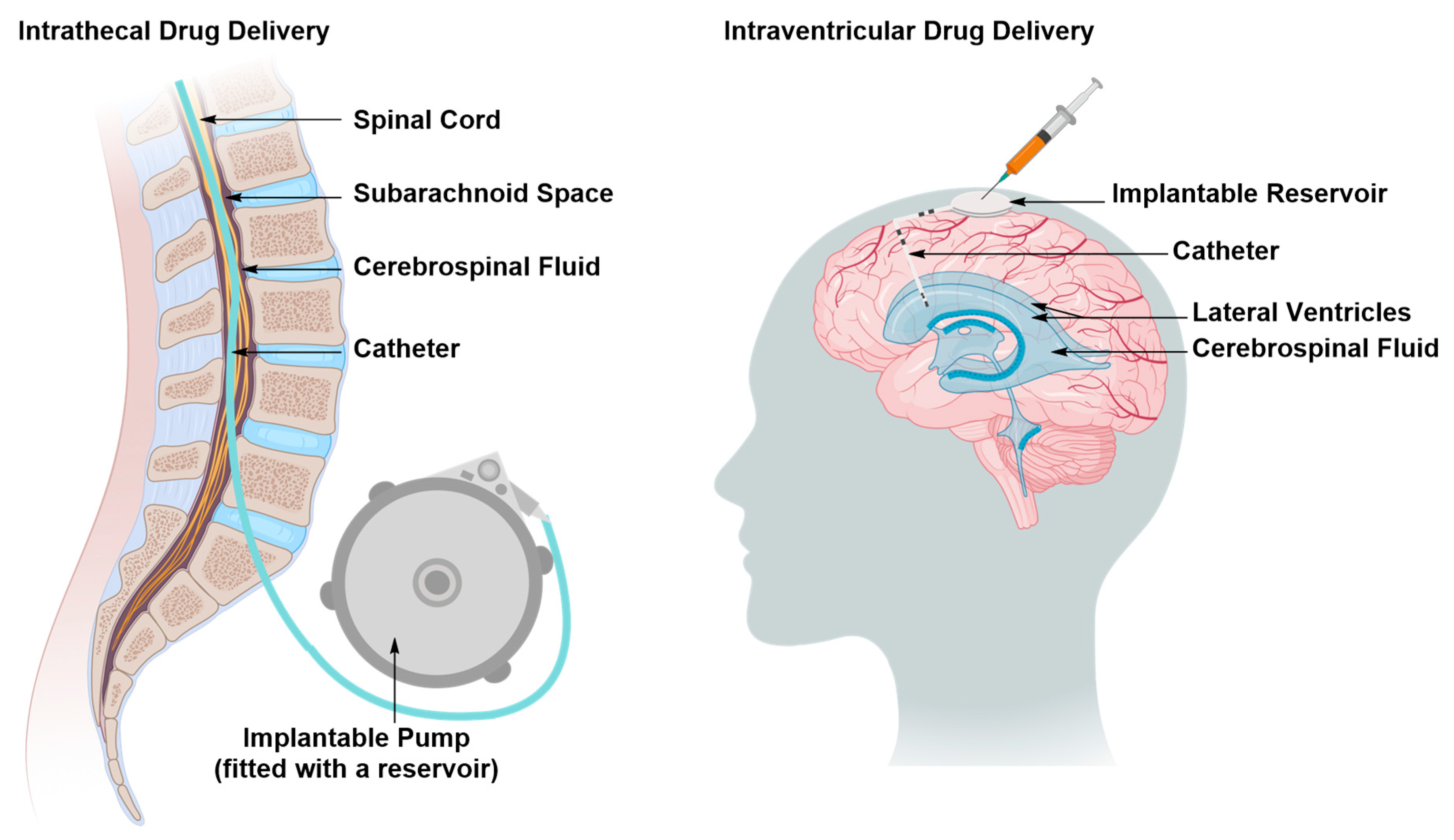
Intraventricular and Intrathecal Drug Delivery
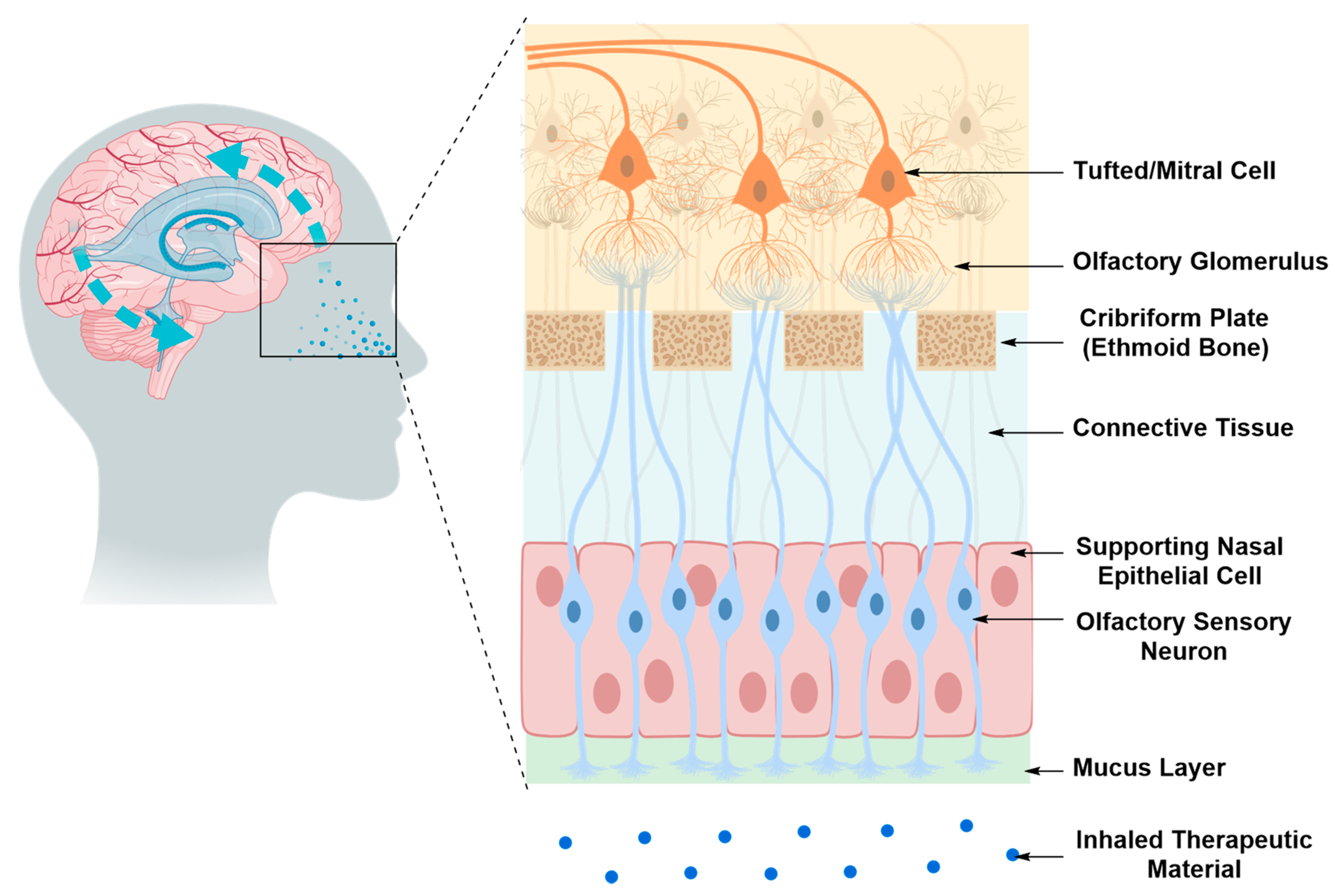
Intranasal Drug Delivery
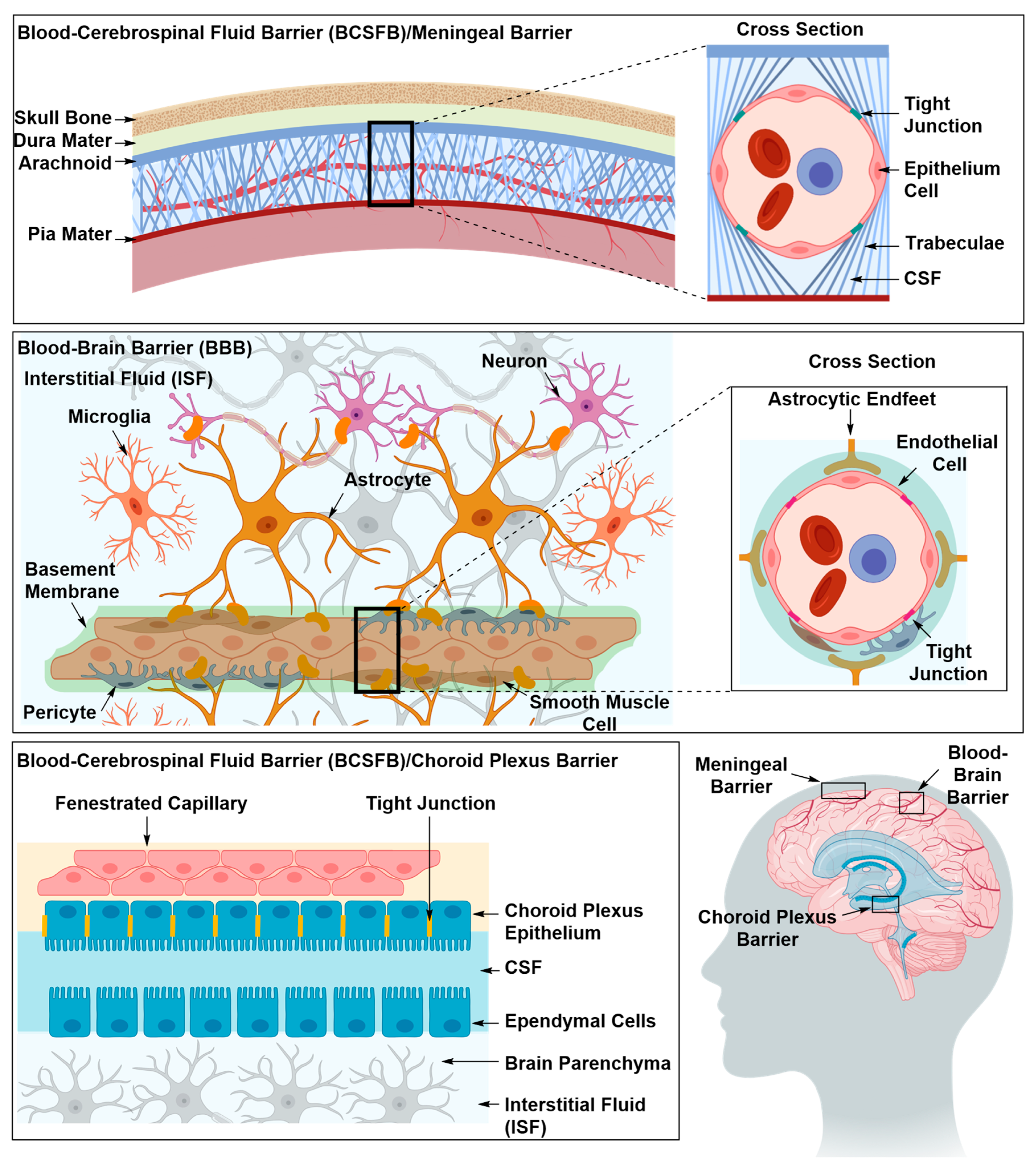
2.1.2. Drug Delivery Through the BBB
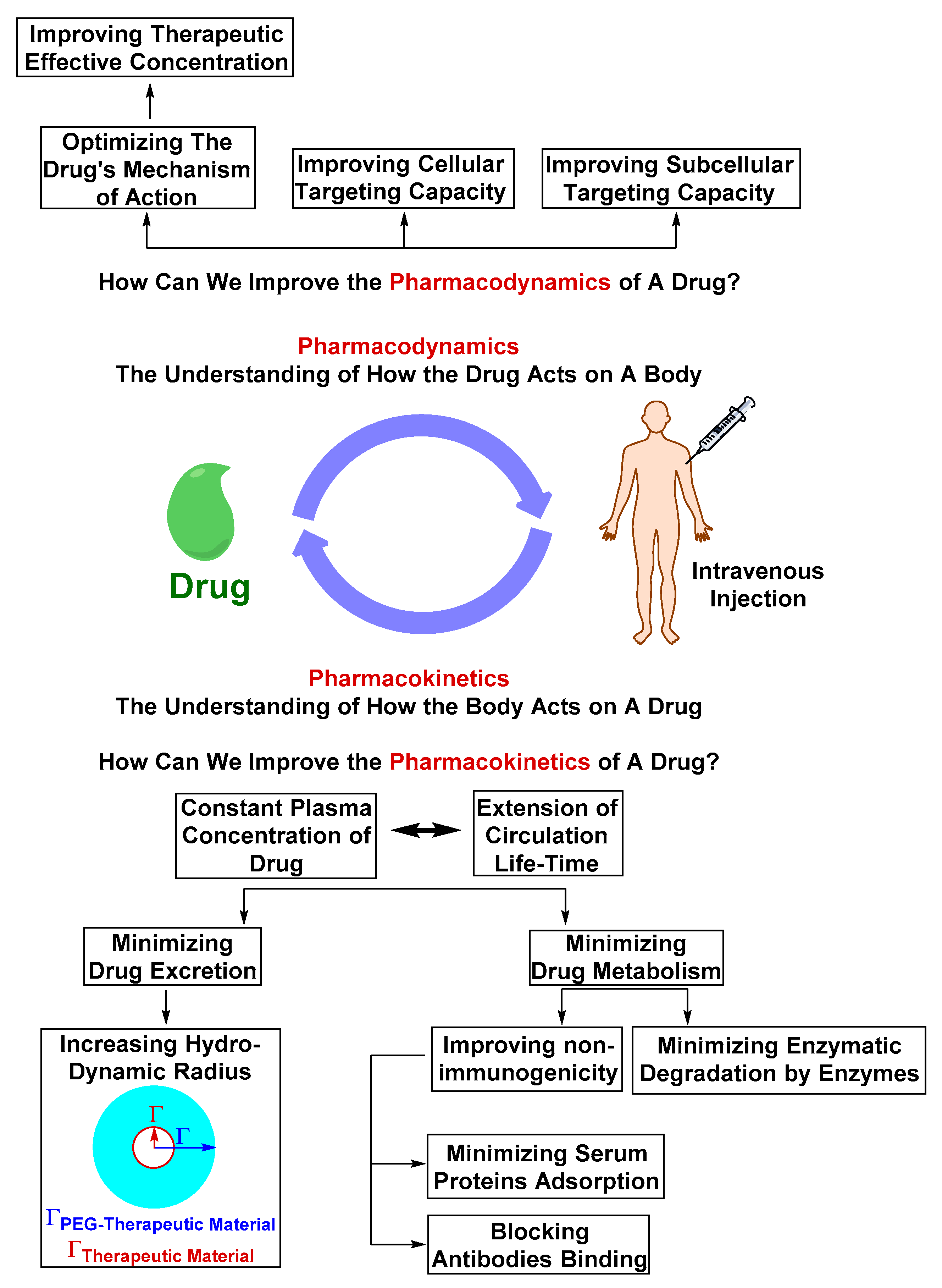
Optimizing the Physicochemical Properties of Therapeutic Materials
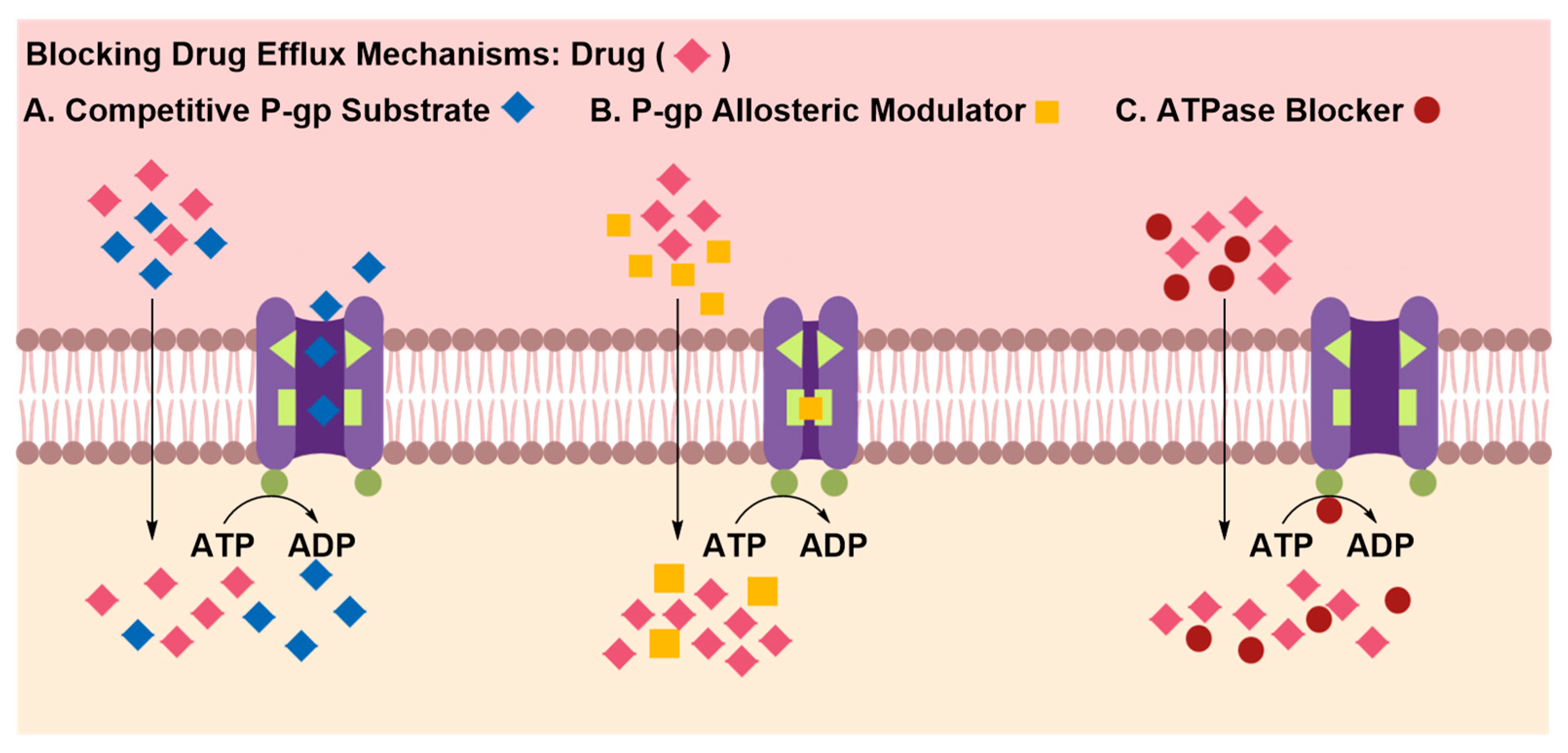
Inhibition of Efflux Proteins
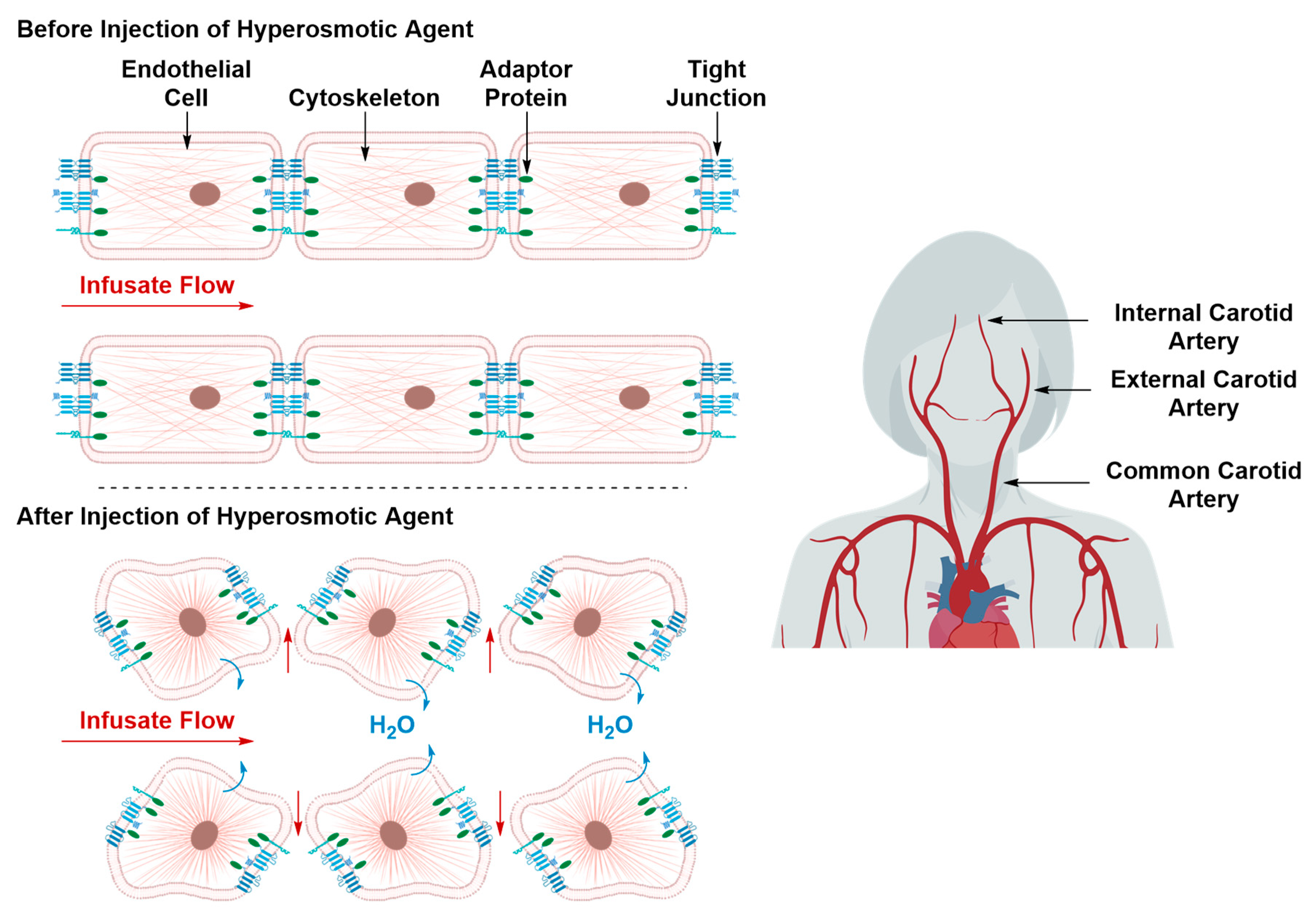
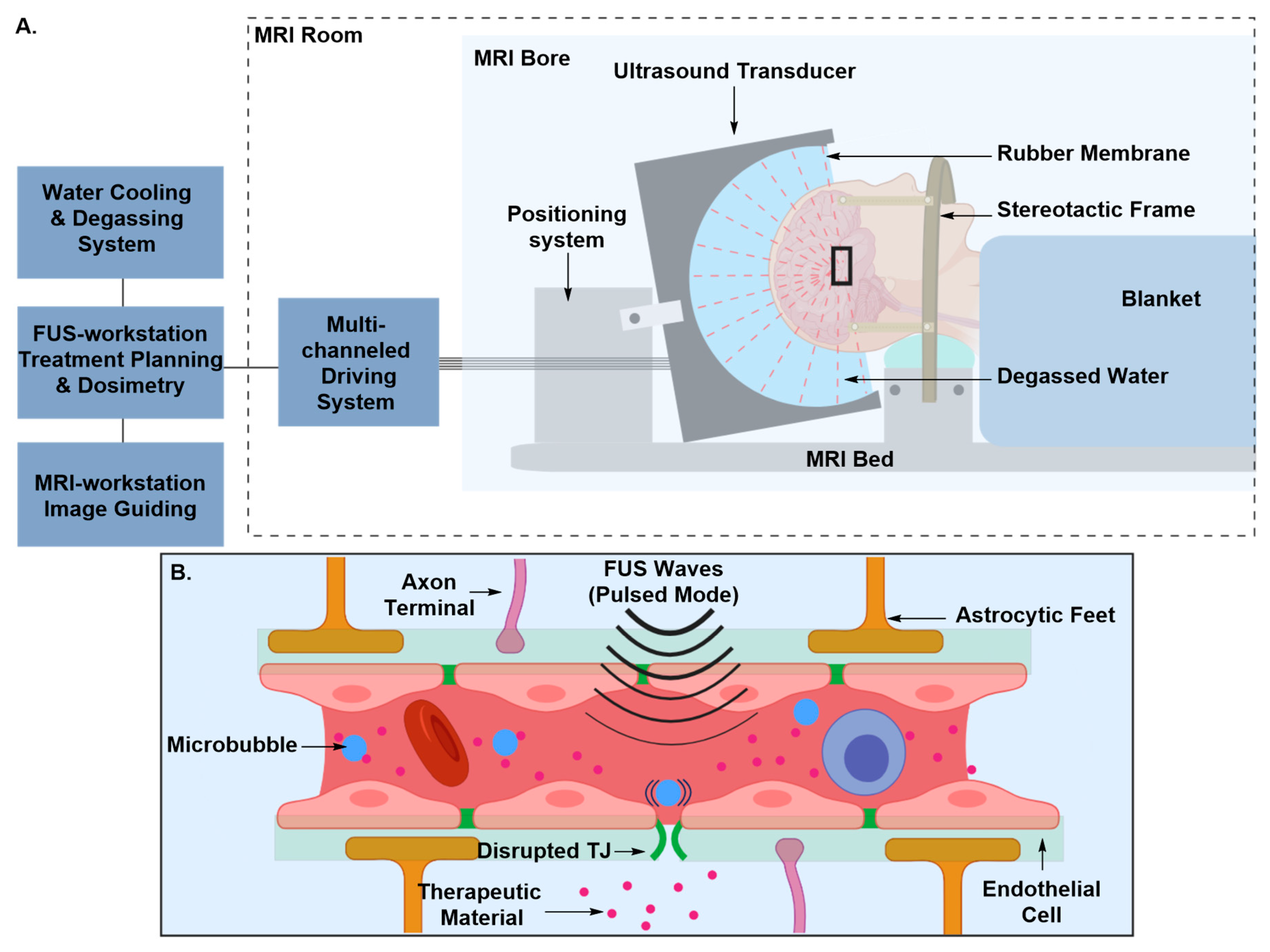
Transient Disruption of the Blood–Brain Barrier
2.1.3. Drug Delivery Using Novel Formulations
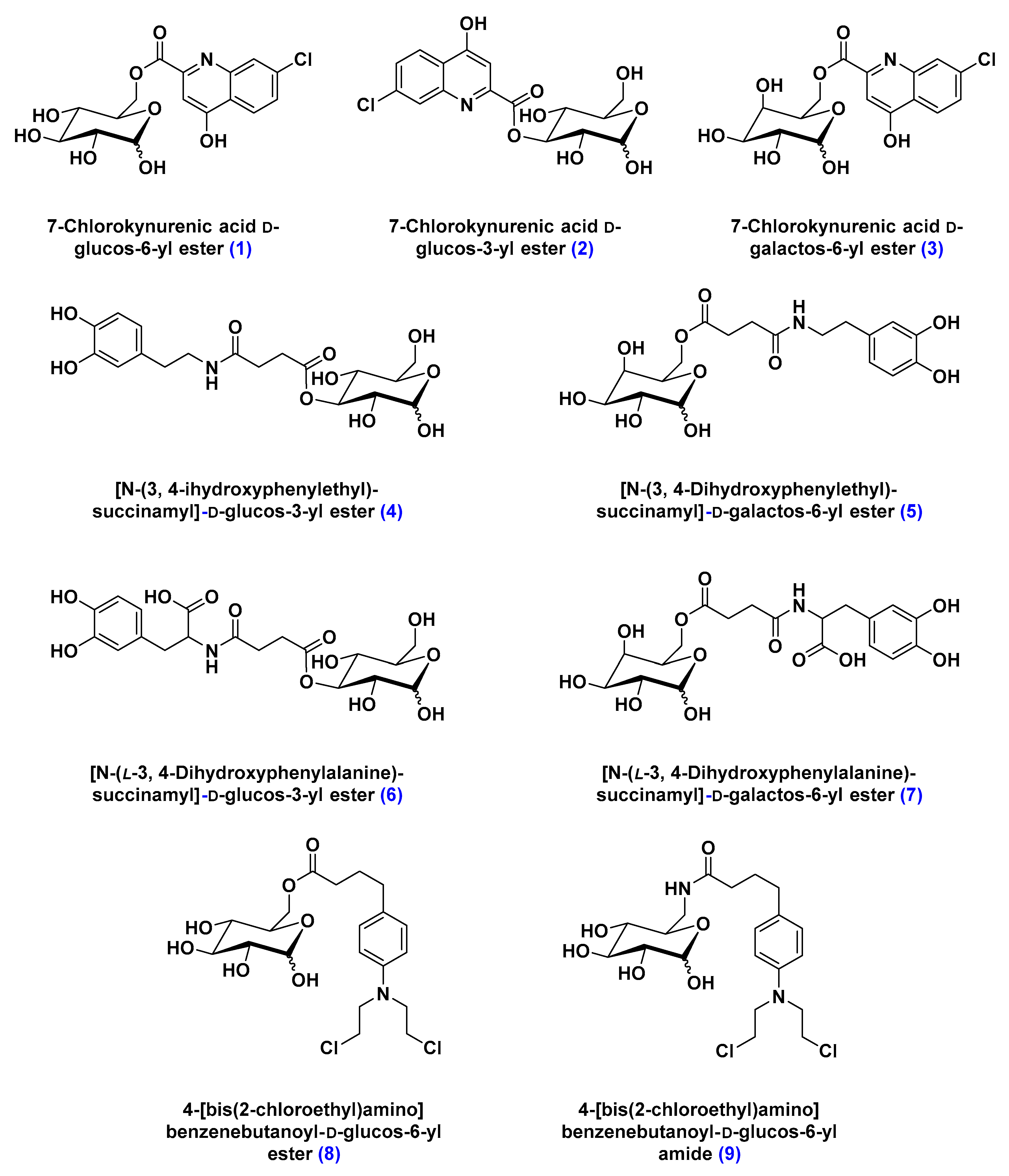
Solute Carrier-Mediated Transcytosis (CMT)
Glucose Transporters (GLUTs)
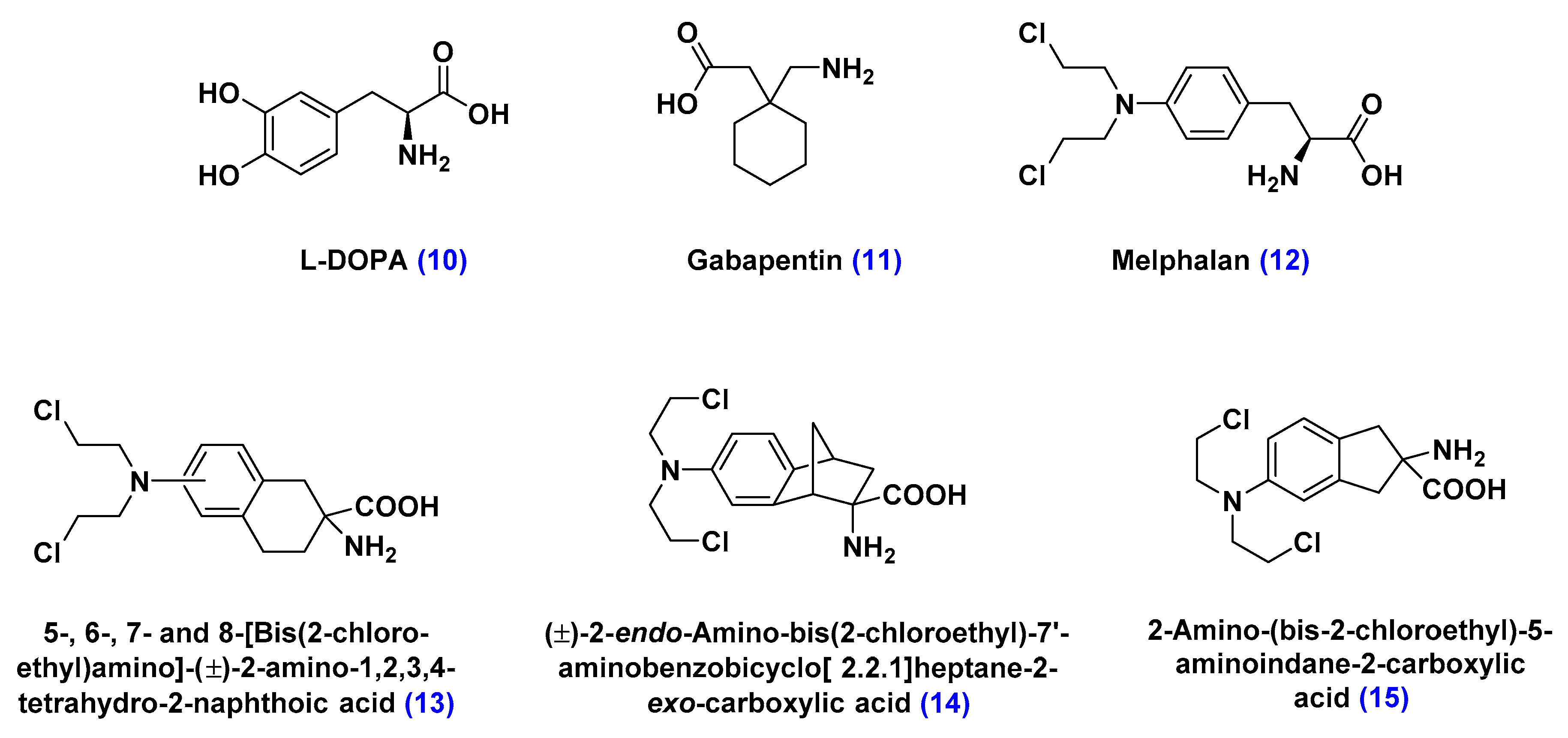
Large Neutral Amino Acid Transporters (LATs)
Organic Anion Transporting Polypeptide (OATPs)
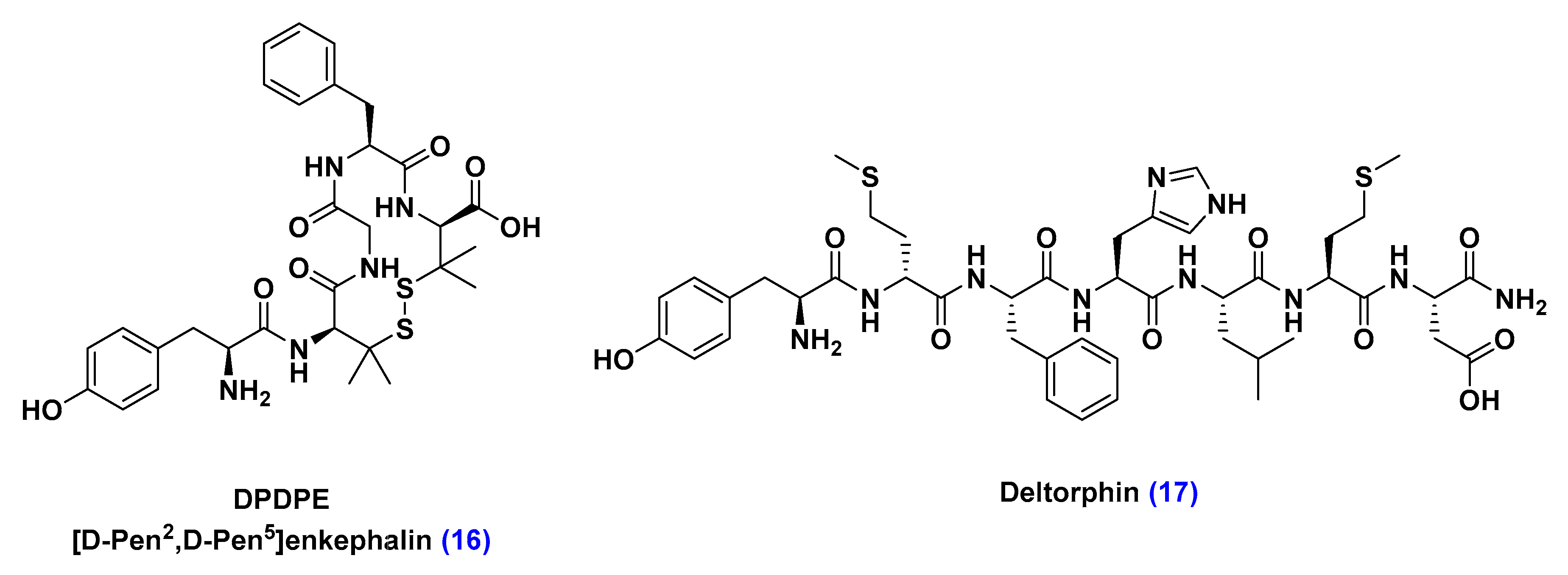
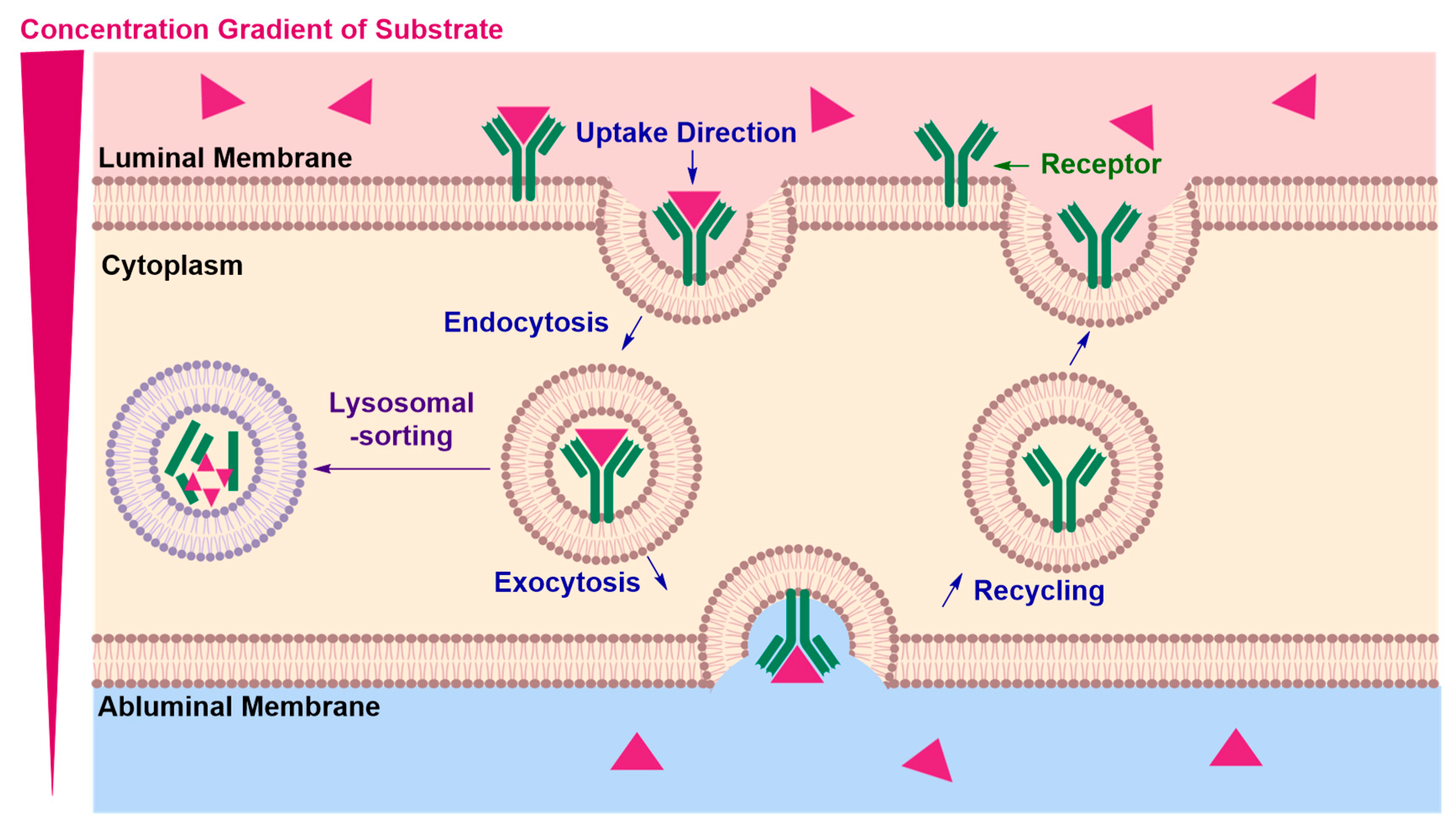
Receptor-Mediated Transcytosis (RMT)
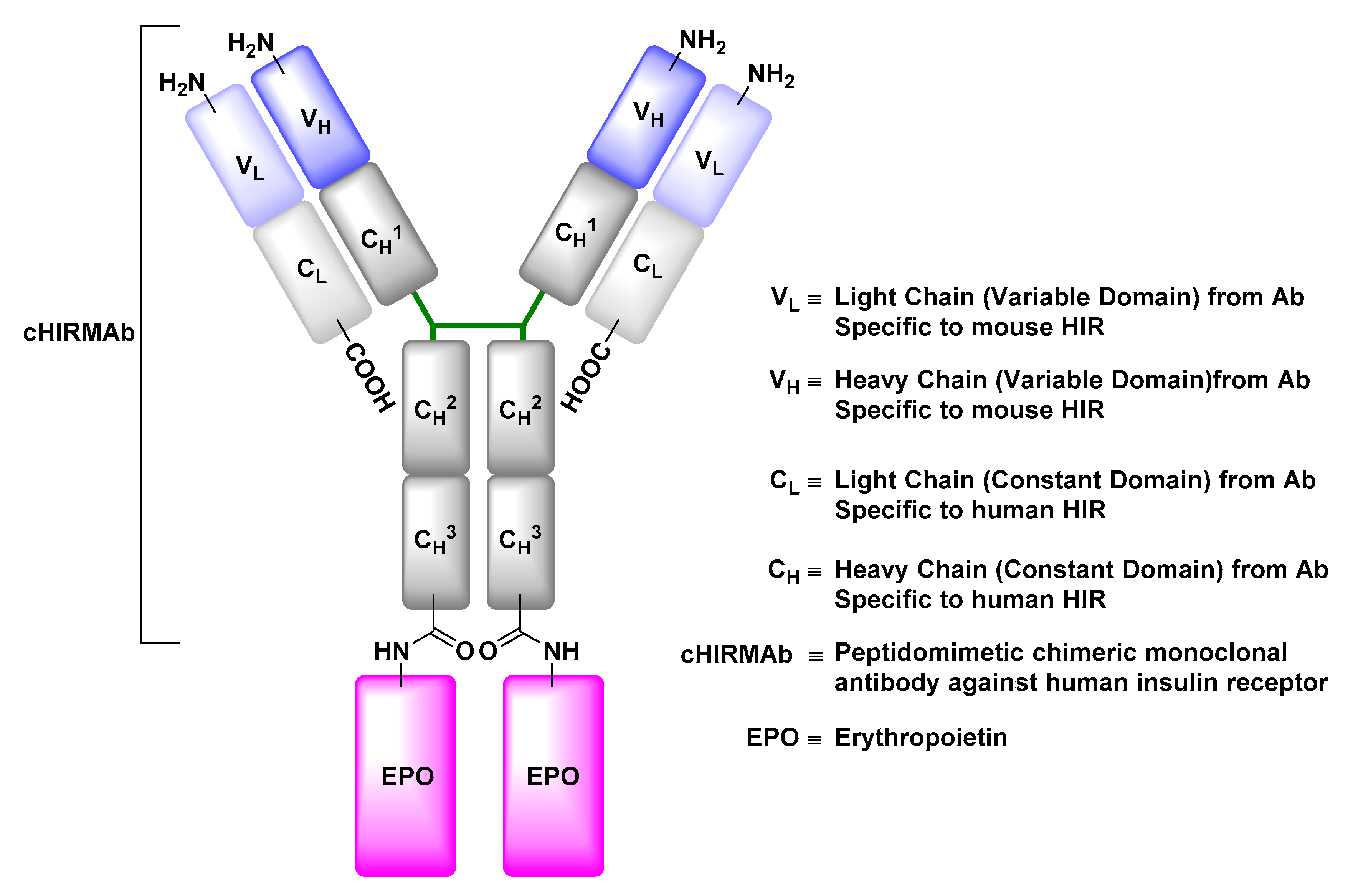
Insulin Receptor (IR)
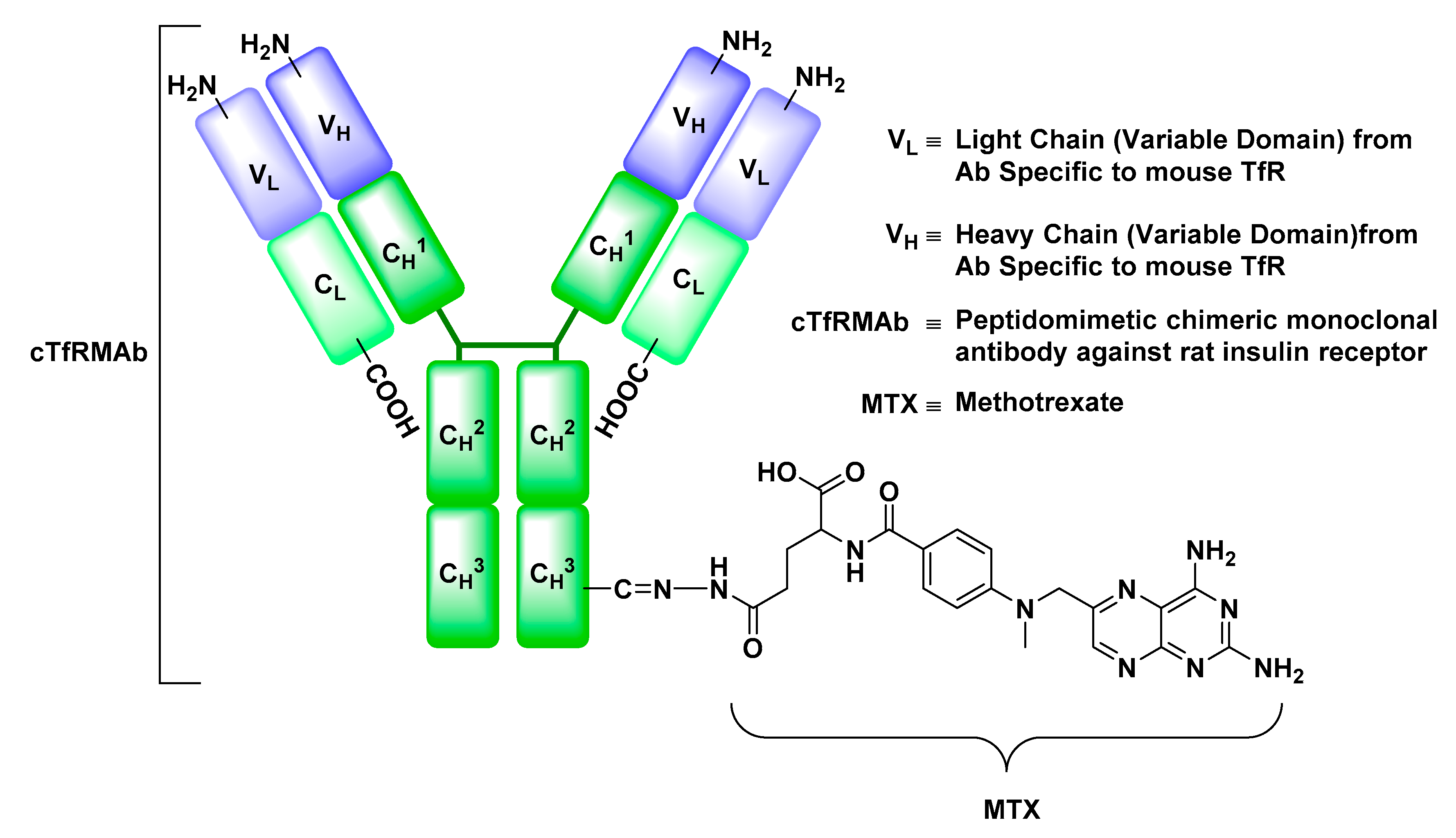
Transferrin Receptor (TfR)
Low-Density Lipoprotein Receptor (LDLR) and Low-Density Lipoprotein Receptor-Related Protein (LPR)
Neurotropic Virulence Factor Receptors (VFRs)
Adsorptive-Mediated Transcytosis (AMT)
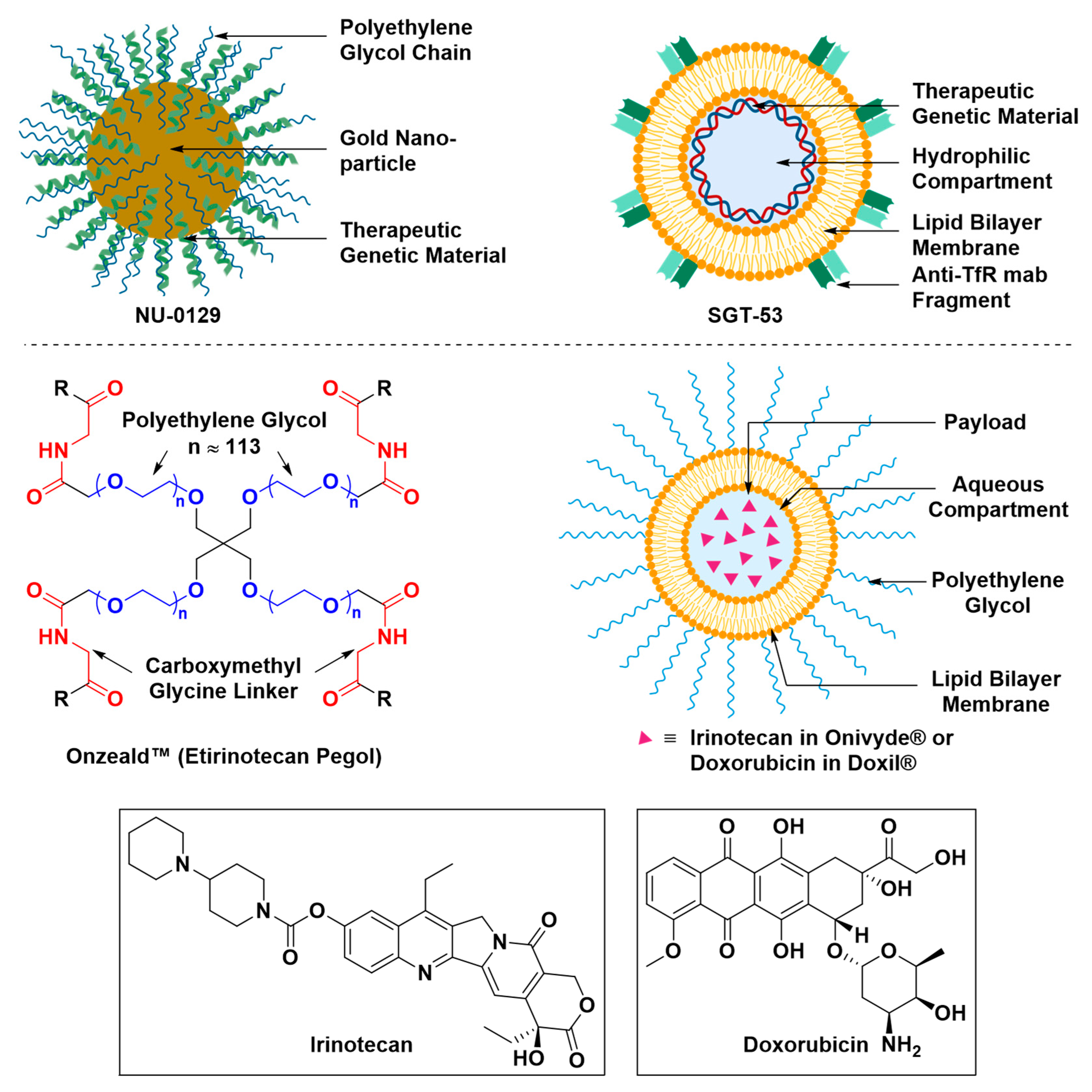
Nanoscopic Formulations for the Delivery of Therapeutics to the Brain
2.2. Challenges in Translation from Pre-clinical Evaluation to Clinical Use
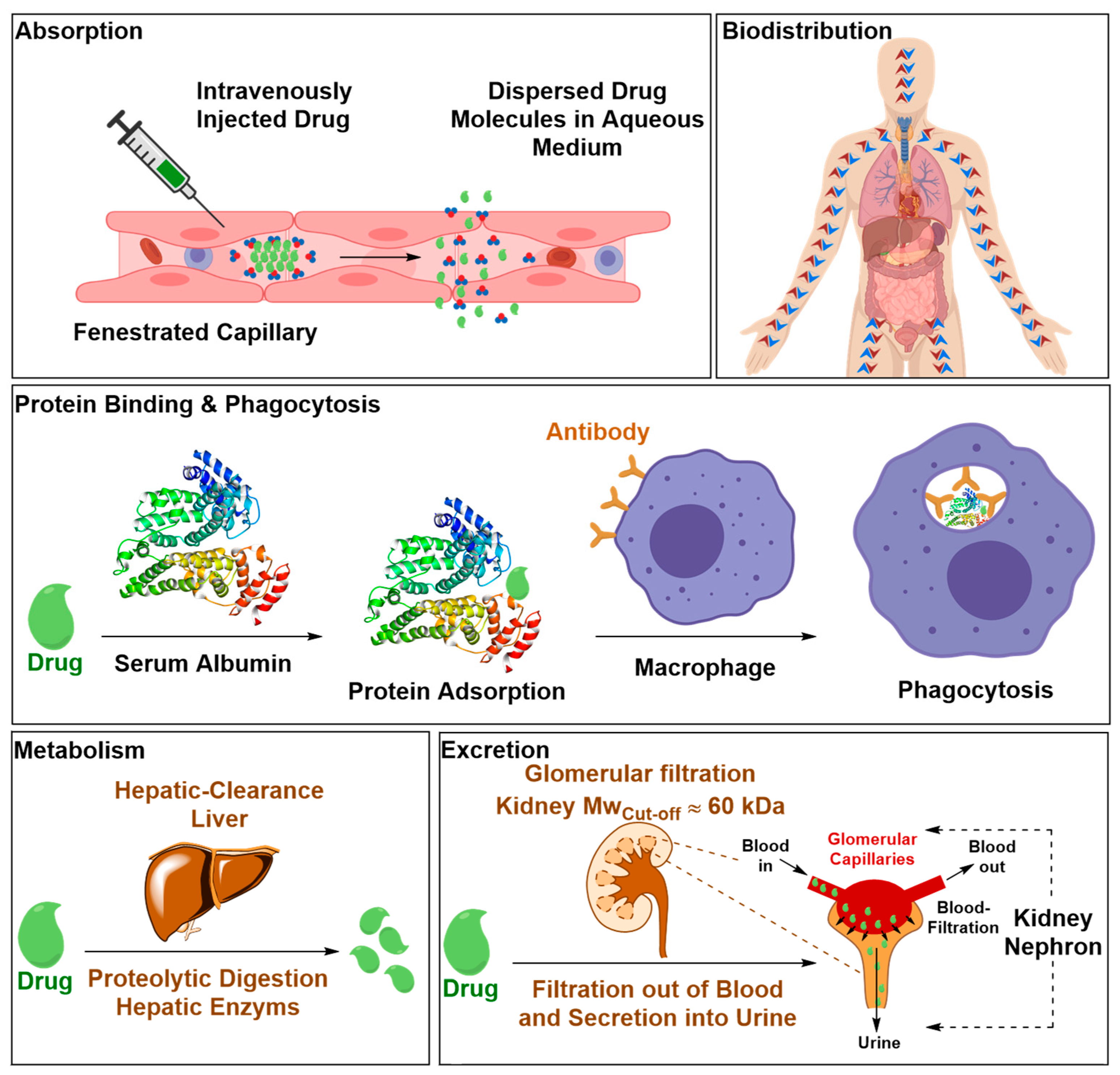
2.2.1. Safety Liabilities due to Nonspecific Body Distribution
2.2.2. Inadequate Endothelial-Parenchymal Transport
2.2.3. How We Measure Drug Transport across the BBB
2.2.4. Species Differ in Parameters that control the BBB permeability
2.2.5. BBB Protein Expression is Altered under Different Pathological Conditions
- How does microvessel composition of carrier proteins, receptors and acidic and neutral lipids in the brain compare to microvessel composition in other major body organs, such as kidneys, liver, spleen and lungs, and how does this vary among different species?
- What is the significance of pathological BBB breakdown in the context of targeted drug delivery? Can BBB breakdown act as a gateway for therapeutic access?
- Is the downregulation of transporters activity a phenotypic hallmark of a dysfunctional BBB?
- How does the alteration of transporters’ activity progress with the aggravation of disease state and how does it vary under different pathological conditions?
- How would advances in the knowledge surrounding pathological BBB breakdown shape the future of research being conducted to exploit the CMT and RMT capacity of ECs in the context of drug delivery through the BBB?
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine Triphosphate |
| AMT | Adsorptive-mediated Transcytosis |
| AD | Alzheimer’s Disease |
| HIR-MAb | Anti-human Insulin Receptor Monoclonal Antibody |
| TfR-MAb | Anti-transferrin Receptor Monoclonal Antibody |
| AUC0T | Area under the Plasma Drug Concentration-time Curve |
| ASL-MRI | Arterial Spin labelling Coupled Magnetic Resonance Imaging |
| ABC | ATP-binding Cassette Transporters |
| BBB | Blood Brain Barrier |
| BCSFB | Blood Cerebrospinal Fluid Barrier |
| Cbr | Brain Concentration |
| BCRP | Breast Cancer Resistance Protein |
| CMT | Carrier-mediated Transcytosis |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| Ð | Dispersity Index |
| DCE-MRI | Dynamic Contrast Enhanced Magnetic Resonance Imaging |
| EC | Endothelial Cell |
| EPO | Erythropoietin |
| GLUT | Glucose Transporter |
| GFP | Green Fluorescent Protein |
| IR | Insulin Receptor |
| ISF | Interstitial Fluid |
| l-DOPA | l-3,4-dihydroxyphenylalanine |
| LAT | Large Neutral Amino Acid Transporter |
| LBL | Lipid Bilayer |
| LRP | Lipoprotein Receptor-related Protein |
| LC-MS/MS | Liquid Chromatography Coupled Mass Spectrometry |
| LDLR | Low-density Lipoprotein Receptor |
| MB-FUS | Microbubble-assisted Focused Ultrasound |
| MCT | Monocarboxylate Transporter |
| MRP | Multidrug Resistance-associated Protein |
| NPBA | Nanoscopic Polymeric Brush Architectures |
| NMDA | N-methyl-D-aspartate |
| Mn | Number Average Molecular Weight |
| OTAP | Organic-anion-transporting Polypeptide |
| PD | Parkinson Disease |
| P-gp | P-glycoprotein |
| PET | Positron-emission Tomography |
| QTAP | Quantitative-targeted Absolute Proteomics |
| RV | Rabies Virus |
| RVG | Rabies Virus Glycoprotein |
| RMT | Receptor-mediated Transcytosis |
| ROMP | Ring Opening Metathesis Polymerization |
| siRNA | Small Interfering Ribonucleic Acid |
| SLC | Solute Carrier Transporter Proteins |
| TJ | Tight Junction |
| TfR | Transferrin Receptor |
| FDA | U S Food and Drug Administration |
| VFR | Virulence Factor Receptor |
References
- Ruttala, H.B.; Ramasamy, T.; Poudal, B.K.; Choi, Y.; Choi, J.Y.; Kim, J.; Kwang Ku, S.; Choi, H.G.; Soon Yong, C.; Oh Kim, J. Molecularly targeted co-delivery of a histone deacetylase inhibitor and paclitaxel by lipid-protein hybrid nanoparticles for synergistic combinational chemotherapy. Oncotarget 2017, 8, 14925–14940. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Ding, J.; He, C.; Cui, L.; Tang, C.; Yin, C. Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 2009, 30, 5691–5700. [Google Scholar] [CrossRef] [PubMed]
- Roney, C.; Kulkarni, P.; Arora, V.; Antich, P.; Bonte, F.; Wu, A.; Mallikarjuana, N.N.; Manohar, S.; Liang, H.F.; Kulkarni, A.R.; et al. Targeted nanoparticles for drug delivery through the blood–brain barrier for Alzheimer’s disease. J. Control. Release 2005, 108, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Shargel, L.; Wu-Pong, S.; Yu, A.B.C. Drug elimination and clearance. In Applied Biopharmaceutics & Pharmacokinetics; McGraw Hill Professional: New York, NY, USA, 2005; pp. 96–116. [Google Scholar]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Ek, C.J.; Habgood, M.D.; Dziegielewska, K.M. Barriers in the brain: A renaissance? Trends Neurosci. 2008, 31, 279–286. [Google Scholar] [CrossRef]
- De Bock, M.; Vandenbroucke, R.E.; Decrock, E.; Culot, M.; Cecchelli, R.; Leybaert, L. A new angle on blood-CNS interfaces: A role for connexins? FEBS Lett. 2014, 588, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- Stock, A.D.; Gelb, S.; Pasternak, O.; Ben-Zvi, A.; Putterman, C. The blood brain barrier and neuropsychiatric lupus: New perspectives in light of advances in understanding the neuroimmune interface. Autoimmun. Rev. 2017, 16, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Alexandre, P. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, 20412–20435. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood–brain barrier: Development, composition and regulation. Vasc. Pharmacol. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Wang, Z.B.; Zhang, L.C.; Wei, X.; Li, L. Tight junction in blood-brain barrier: An overview of structure, regulation, and regulator substances. CNS Neurosci. Ther. 2012, 18, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer’s disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Maeda, T.; Abe, M.A.; Nakayama, H.; Takahashi, R.; Ueda, M.; Ohtsuki, S.; Terasaki, T.; Obinata, M.; et al. Peripheral nerve pericytes originating from the blood-nerve barrier expresses tight junctional molecules and transporters as barrier-forming cells. J. Cell. Physiol. 2008, 217, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, A.; Tanaka, K.; Niwa, M. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Al-Ahmady, Z.S. Selective drug delivery approaches to lesioned brain through blood brain barrier disruption. Expert Opin. Drug Deliv. 2018, 15, 335–349. [Google Scholar] [CrossRef]
- Saunders, N.R.; Dziegielewska, K.M.; Møllgård, K.; Habgood, M.D. General Introduction to Barrier Mechanisms in the Central Nervous System. In The Blood Brain Barrier and Inflammation; Lyck, R., Enzmann, G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–22. [Google Scholar]
- Mikitsh, J.L.; Chacko, A.M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Med. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Mokgokong, R.; Wang, S.; Taylor, C.J.; Barrand, M.A.; Hladky, S.B. Ion transporters in brain endothelial cells that contribute to formation of brain interstitial fluid. Pflugers Arch. 2014, 466, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Patel, A.; Kathawala, R.J.; Chen, Z.S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58–72. [Google Scholar] [CrossRef]
- Varma, M.V.S.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 2003, 48, 347–359. [Google Scholar] [CrossRef]
- Raghavan, R. Intraparenchymal Delivery and Its Discontents. In Drug Delivery to the Central Nervous System; Jain, K.K., Ed.; Humana Press: New York, NY, USA, 2010; pp. 85–135. [Google Scholar]
- Hladky, S.B.; Barrand, M.A. Mechanisms of fluid movement into, through and out of the brain: Evaluation of the evidence. Fluids Barriers CNS 2014, 11, 26–58. [Google Scholar] [CrossRef]
- Lei, Y.; Han, H.; Yuan, F.; Javeed, A.; Zhao, Y. The brain interstitial system: Anatomy, modeling, in vivo measurement, and applications. Prog. Neurobiol. 2017, 157, 230–246. [Google Scholar] [CrossRef]
- Belverud, S.; Mogilner, A.; Schulder, M. Intrathecal drug delivery by implanted pumps. In Drug Delivery to the Central Nervous System; Jain, K.K., Ed.; Humana Press: New York, NY, USA, 2010; pp. 137–153. [Google Scholar]
- Dossani, R.H.; Kalakoti, P.; Thakur, J.D.; Nanda, A. Ayub Khan Ommaya (1930–2008): Legacy and contributions to neurosurgery. Neurosurgery 2017, 80, 324–330. [Google Scholar] [CrossRef]
- Cook, A.M.; Mieure, K.D.; Owen, R.D.; Pesaturo, A.B.; Hatton, J. Intracerebroventricular administration of drugs. Pharmacotherapy 2009, 29, 832–845. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, T.M. Direct intraventricular delivery of drugs to the rodent central nervous system. J. Vis. Exp. 2013, 75, 50326–50336. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, M.J.R.; de Lange, E.C.M. Emerging insights for translational pharmacokinetic and Pharmacokinetic-pharmacodynamic studies: Towards prediction of nose-to-brain transport in humans. AAPS J. 2015, 17, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Illum, L. Nasal drug delivery—Recent developments and future prospects. J. Control. Release 2012, 161, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Lanevskij, K.; Japertas, P.; Didziapetris, R.; Petrauskas, A. Prediction of blood–brain barrier penetration by drugs. In Drug Delivery to the Central Nervous System; Jain, K.K., Ed.; Humana Press: New York, NY, USA, 2010; pp. 63–83. [Google Scholar]
- Bickel, U. How to measure drug transport across the blood-brain barrier. NeuroRx 2005, 2, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.B.; Segretti, M.C.F.; Polli, M.C.; Parise-Filho, R. Analysis of the applicability and use of lipinski’s rule for central nervous system drugs. Lett. Drug Des. Discov. 2016, 13, 999–1006. [Google Scholar] [CrossRef]
- Banks, W.A. From blood–brain barrier to blood– brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Crawford, L.; Rosch, J.; Putnam, D. Concepts, technologies, and practices for drug delivery past the blood-brain barrier to the central nervous system. J. Control. Release 2016, 240, 251–266. [Google Scholar] [CrossRef]
- Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef]
- Freire, E. The binding thermodynamics of drug candidates. In Thermodynamics and Kinetics of Drug Binding; Keserü, G.M., Swinney, D.C., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2015; pp. 3–13. [Google Scholar]
- Atkovska, K.; Klingler, J.; Oberwinkler, J.; Keller, S. Rationalizing steroid interactions with lipid membranes: Conformations, partitioning, and kinetics. ACS Cent. Sci. 2018, 4, 1155–1165. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Parrish, K.E.; Sarkaria, J.N.; Elmquist, W.F. Improving drug delivery to primary and metastatic brain tumors: Strategies to overcome the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Kemper, E.M.; van Zandbergen, A.E.; Cleypool, C.; Mos, H.A.; Boogerd, W.; Beijnen, J.H.; van Tellingen, O. Increased penetration of paclitaxel into the brain by inhibition of P-Glycoprotein. Clin. Cancer Res. 2003, 9, 2849–2855. [Google Scholar] [PubMed]
- Srivalli, K.M.R.; Lakshmi, P.K. Overview of P-glycoprotein inhibitors: A rational outlook. Braz. J. Pharm. Sci. 2012, 48, 353–367. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I. Osmotic opening of the blood–brain barrier: Principles, mechanism, and therapeutic applications. Cell. Mol. Neurobiol. 2000, 20, 217–230. [Google Scholar] [CrossRef]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving drug delivery strategies to overcome the blood brain barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef]
- McDannold, N.; Clement, G.; Black, P.; Jolesz, F.; Hynynen, K. Transcranial MRI-guided focused ultrasound surgery of brain tumors: Initial findings in three patients. Neurosurgery 2010, 66, 323–332. [Google Scholar] [CrossRef]
- Martin, E.; Werner, B. Focused ultrasound surgery of the brain. Curr. Radiol. Rep. 2013, 1, 126–135. [Google Scholar] [CrossRef]
- Iacopino, D.G.; Gagliardo, C.; Giugno, A.; Giammalva, G.R.; Napoli, A.; Maugeri, R.; Graziano, F.; Valentino, F.; Cosentino, G.; D’Amelio, M.; et al. Preliminary experience with a transcranial magnetic resonance-guided focused ultrasound surgery system integrated with a 1.5-T MRI unit in a series of patients with essential tremor and Parkinson’s disease. Neurosurg. Focus 2018, 44, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J.T.; Nolting, M.N.; Goss, S.A.; Dines, K.A.; Clendenon, J.L.; Rea, M.A.; Heimburger, R.F. Ultrasound and the blood-brain barrier. In Consensus on Hyperthermia for the 1990s; Bicher, H.I., McLaren, J.R., Pigliucci, G.M., Eds.; Springer: Boston, MA, USA, 1990; pp. 369–381. [Google Scholar]
- Vykhodtseva, N.I.; Hynynen, K.; Damianou, C. Histologic effects of high intensity pulsed ultrasound exposure with subharmonic emission in rabbit brain in vivo. Ultrasound Med. Biol. 1995, 21, 969–979. [Google Scholar] [CrossRef]
- Mesiwala, A.H.; Farrell, L.; Wenzel, H.J.; Silbergeld, D.L.; Crum, L.A.; Winn, H.R.; Mourad, P.D. High-intensity focused ultrasound selectively disrupts the blood-brain barrier in vivo. Ultrasound Med. Biol. 2002, 28, 389–400. [Google Scholar] [CrossRef]
- McDannold, N.; Vykhodtseva, N.; Jolesz, F.A.; Hynynen, K. MRI investigation of the threshold for thermally induced blood-brain barrier disruption and brain tissue damage in the rabbit brain. Magn. Reson. Med. 2004, 51, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Aryal, M.; Arvanitis, C.D.; Alexander, P.M.; McDannold, N. Ultrasound-mediated blood-brain barrier disruption for targeted drug delivery in the central nervous system. Adv. Drug Deliv. Rev. 2014, 72, 94–109. [Google Scholar] [CrossRef]
- Vykhodtseva, N. Disruption of blood–brain barrier by focused ultrasound for targeted drug delivery to the brain. In Drug Delivery to the Central Nervous System; Jain, K.K., Ed.; Humana Press: New York, NY, USA, 2010; pp. 35–62. [Google Scholar]
- Park, J.; Zhang, Y.; Vykhodtseva, N.; Akula, J.D.; McDannold, N.J. Targeted and reversible blood-retinal barrier disruption via focused ultrasound and microbubbles. PLoS ONE 2012, 7, e42754. [Google Scholar] [CrossRef]
- Shi, L.; Palacio-Mancheno, P.; Badami, J.; Shin, D.W.; Zeng, M.; Cardoso, L.; Tu, R.; Fu, B.M. Quantification of transient increase of the blood–brain barrier permeability to macromolecules by optimized focused ultrasound combined with microbubbles. Int. J. Nanomed. 2014, 9, 4437–4448. [Google Scholar]
- Sheikov, N.; McDannold, N.; Sharma, S.; Hynynen, K. Effect of focused ultrasound applied with an ultrasound contrast agent on the tight junctional integrity of the brain microvascular endothelium. Ultrasound Med. Biol. 2008, 34, 1093–1104. [Google Scholar] [CrossRef]
- Kinoshita, M.; McDannold, N.; Jolesz, F.A.; Hynynen, H.K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proc. Natl. Acad. Sci. USA 2006, 103, 11719–11723. [Google Scholar] [CrossRef]
- Treat, L.H.; McDannold, N.; Vykhodtseva, N.; Zhang, Y.; Tam, K.; Hynynen, K. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. Int. J. Cancer 2007, 121, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Aubry, J.F.; Tanter, M.; Pernot, M.; Thomas, J.L.; Fink, M. Experimental demonstration of noninvasive transskull adaptive focusing based on prior computed tomography scans. J. Acoust. Soc. Am. 2003, 113, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Dobrogowska, D.H.; Vorbrodt, A.W. Immunogold localization of tight junctional proteins in normal and osmotically-affected rat blood-brain barrier. J. Mol. Histol. 2004, 35, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Sugiyama, Y. Membrane transporters and drug response. In Goodman & Gilman’s The Pharmacological Basis of Therapeutics; Brunton, L.L., Lazo, J.S., Parker, K.L., Eds.; McGraw-Hill Professional: New York, NY, USA, 2005; pp. 41–69. [Google Scholar]
- Bai, X.; Moraes, T.F.; Reithmeier, R.A.F. Structural biology of solute carrier (SLC) membrane transport proteins. Mol. Membr. Biol. 2017, 34, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef] [PubMed]
- Deo, A.K.; Theil, F.P.; Nicolas, J.M. Confounding parameters in preclinical assessment of blood-brain barrier permeation: An overview with emphasis on species differences and effect of disease states. Mol. Pharm. 2013, 10, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Kamiie, J.; Ohtsuki, S.; Iwase, R.; Ohmine, K.; Katsukura, Y.; Yanai, K.; Sekine, Y.; Uchida, Y.; Ito, S.; Terasaki, T. Quantitative atlas of membrane transporter proteins: Development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm. Res. 2008, 25, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; La Russa, M.; Bruno, V.; Arenare, L.; Ippolito, R.; Copani, A.; Bonina, F.; Nicoletti, F. Systematically administered D-glucose conjugates of 7-chlorokynurenic acid are centrally available and exert anticonvulsant activity in rodents. Brain Res. 2000, 860, 149–156. [Google Scholar] [CrossRef]
- Bonina, F.; Arenare, L.; Ippolito, R.; Boatto, G.; Battaglia, G.; Bruno, V.; de Caparariis, P. Synthesis, pharmacokinetics and anticonvulsant activity of 7-chlorokynurenic acid prodrugs. Int. J. Pharm. 2000, 202, 79–88. [Google Scholar] [CrossRef]
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Melisi, D.; Boatto, G.; Nieddu, M.; Calignano, A.; La Rana, G.; De Caprariis, P. Glycosyl derivatives of dopamine and L-dopa as antiparkinson prodrugs: Synthesis, pharmacological activity and in vitro stability studies. J. Drug Target. 2003, 11, 25–36. [Google Scholar] [PubMed]
- Dalpiaz, A.; Filosa, R.; de Caprariis, P.; Conte, G.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Prasad, P.D.; Pavan, B. Molecular mechanism involved in the transport of a prodrug dopamine glycosyl conjugate. Int. J. Pharm. 2007, 336, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Halmos, T.; Santarromana, M.; Antonakis, K.; Scherman, D. Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur. J. Pharmacol. 1996, 318, 477–484. [Google Scholar] [CrossRef]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.J.; Sali, A.; Giacominia, K.M. Structure-based ligand discovery for the large-neutral amino acid transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef] [PubMed]
- Matharu, J.; Oki, J.; Worthen, D.R.; Smith, Q.R.; Crooksa, P.A. Regiospecific and conformationally restrained analogs of melphalan and DL-2-NAM-7 and their affinities for the large neutral amino acid transporter (system LAT1) of the blood–brain barrier. Bioorg. Med. Chem. Lett. 2010, 20, 3688–3691. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Young, D.; Paxton, J.W.; Finlay, G.J.; Wilson, W.R.; Pardridge, W.M. Melphalan penetration of the blood-brain barrier via the neutral amino acid transporter in tumor-bearing brain. Cancer Res. 1992, 52, 138–143. [Google Scholar] [PubMed]
- Westholm, D.E.; Rumbley, J.N.; Salo, D.R.; Rich, T.P.; Anderson, G.W. Organic anion-transporting polypeptides at the blood-brain and blood-cerebrospinal fluid barriers. Curr. Top. Dev. Biol. 2008, 80, 135–170. [Google Scholar]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar] [PubMed]
- Ronaldson, P.T.; Finch, J.D.; Demarco, K.M.; Quigley, C.E.; Davis, T.P. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J. Pharmacol. Exp. Ther. 2011, 336, 827–839. [Google Scholar] [CrossRef]
- Tuma, P.; Hubbard, A.L. Transcytosis: Crossing Cellular Barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [CrossRef]
- Begg, D.P. Insulin transport into the brain and cerebrospinal fluid. Vitam. Horm. 2015, 98, 229–248. [Google Scholar]
- Bunn, H.F. Erythropoietin. Cold Spring Harb. Perspect. Med. 2013, 3, 11619–11639. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Hui, E.K.; Lu, J.Z.; Pardridge, W.M. Drug targeting of erythropoietin across the primate blood-brain barrier with an IgG molecular Trojan horse. J. Pharmacol. Exp. Ther. 2010, 333, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Pardridge, W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Morgan, E.H. Transferrin and transferrin receptor function in brain barrier systems. Cell. Mol. Neurobiol. 2000, 20, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Friden, P.M.; Walus, L.R.; Musso, G.F.; Taylor, M.A.; Malfroy, B.; Starzyk, R.M. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1991, 88, 4771–4775. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Hui, E.K.; Lu, J.Z.; Boado, R.J.; Pardridge, W.M. Neuroprotection in stroke in the mouse with intravenous erythropoietin-Trojan horse fusion protein. Brain Res. 2011, 1369, 203–207. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Woldt, E.; Matz, R.L.; Boucher, P. The LDL receptor-related protein (LRP) family: An old family of proteins with new physiological functions. Ann. Med. 2007, 39, 219–228. [Google Scholar] [CrossRef] [PubMed]
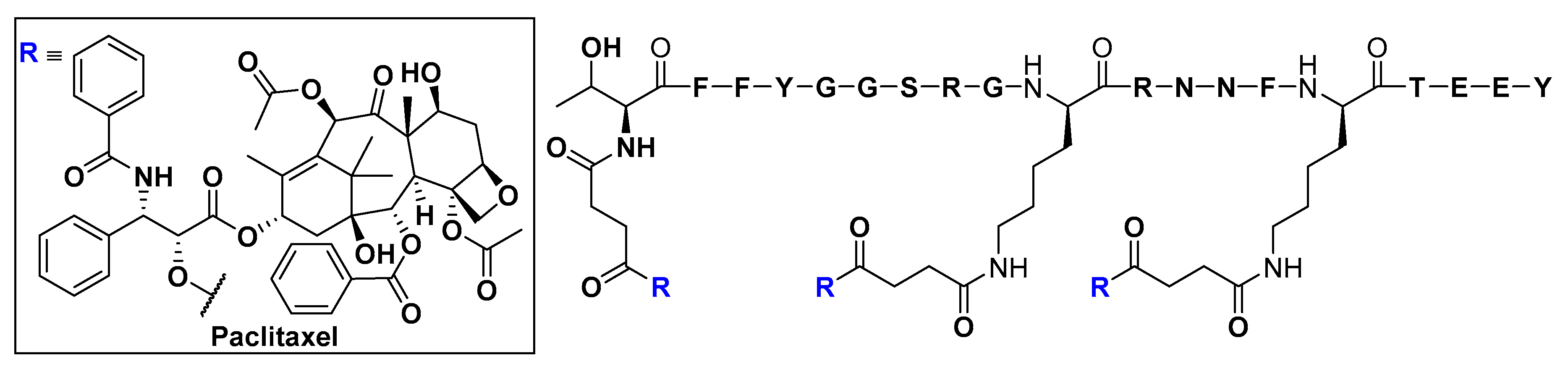
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- Thomas, F.C.; Taskar, K.; Rudraraju, V.; Goda, S.; Thorsheim, H.R.; Gaasch, J.A.; Mittapalli, R.K.; Palmieri, D.; Steeg, P.S.; Lockman, P.R.; et al. Uptake of ANG1005, a novel paclitaxel derivative, through the blood-brain barrier into brain and experimental brain metastases of breast cancer. Pharm. Res. 2009, 26, 2486–2494. [Google Scholar] [CrossRef]
- Cross, A.S. What is a virulence factor? Crit. Care 2008, 12, 196–198. [Google Scholar] [CrossRef]
- Oswald, M.; Geissler, S.; Goepferich, A. Targeting the central nervous system (CNS): A review of rabies virus-targeting strategies. Mol. Pharm. 2017, 14, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Future Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef] [PubMed]
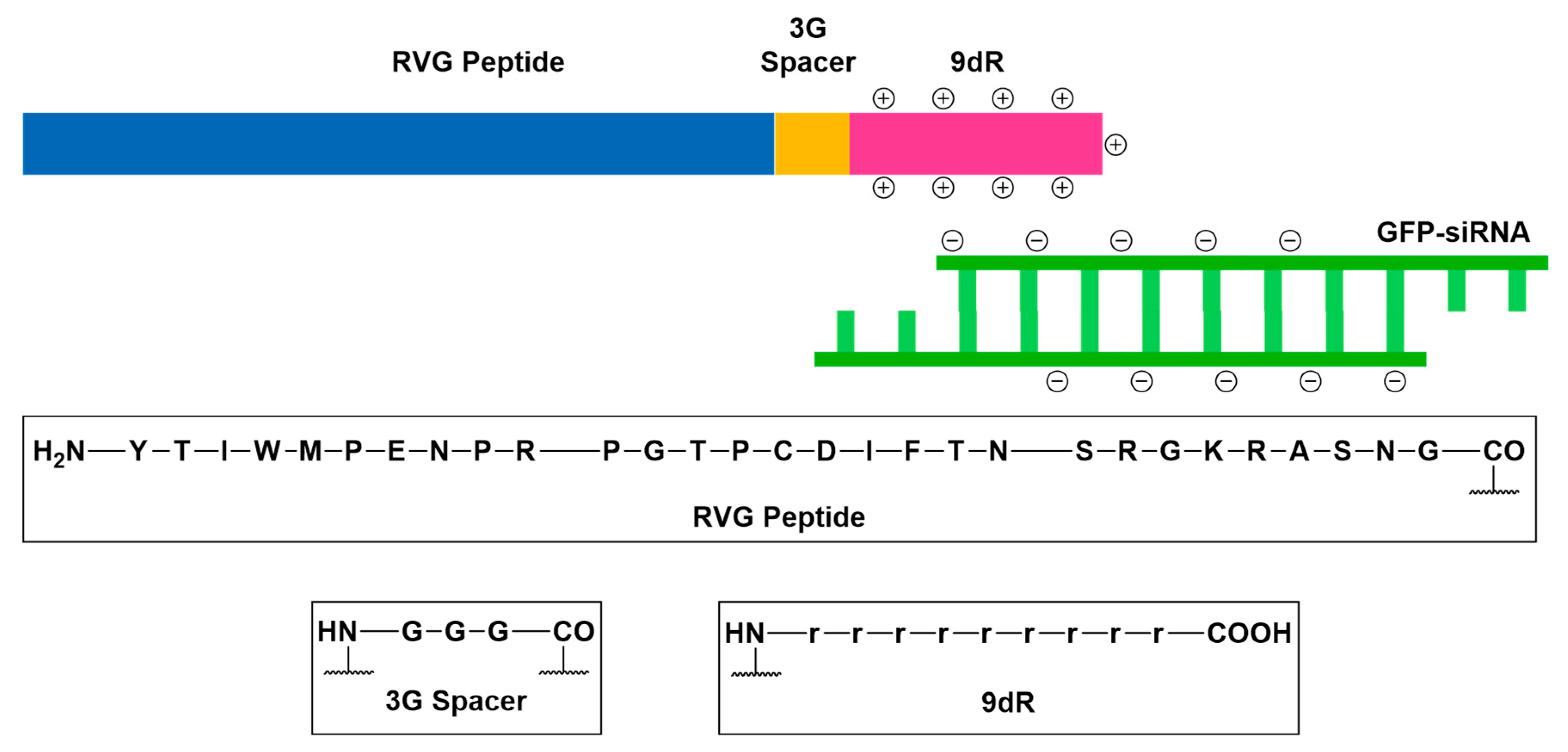
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.J.; McGettigan, J.P.; Wirblich, C.; Papaneri, A. The cell biology of rabies virus: Using stealth to reach the brain. Nat. Rev. Microbiol. 2010, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Hawthorne, S.; McCarron, P. The potential use of rabies virus glycoprotein-derived peptides to facilitate drug delivery into the central nervous system: A mini review. J. Drug Target. 2017, 25, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef]
- Gao, H.; Pang, Z.; Jiang, X. Targeted delivery of nano-therapeutics for major disorders of the central nervous system. Pharm. Res. 2013, 30, 2485–2498. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed]
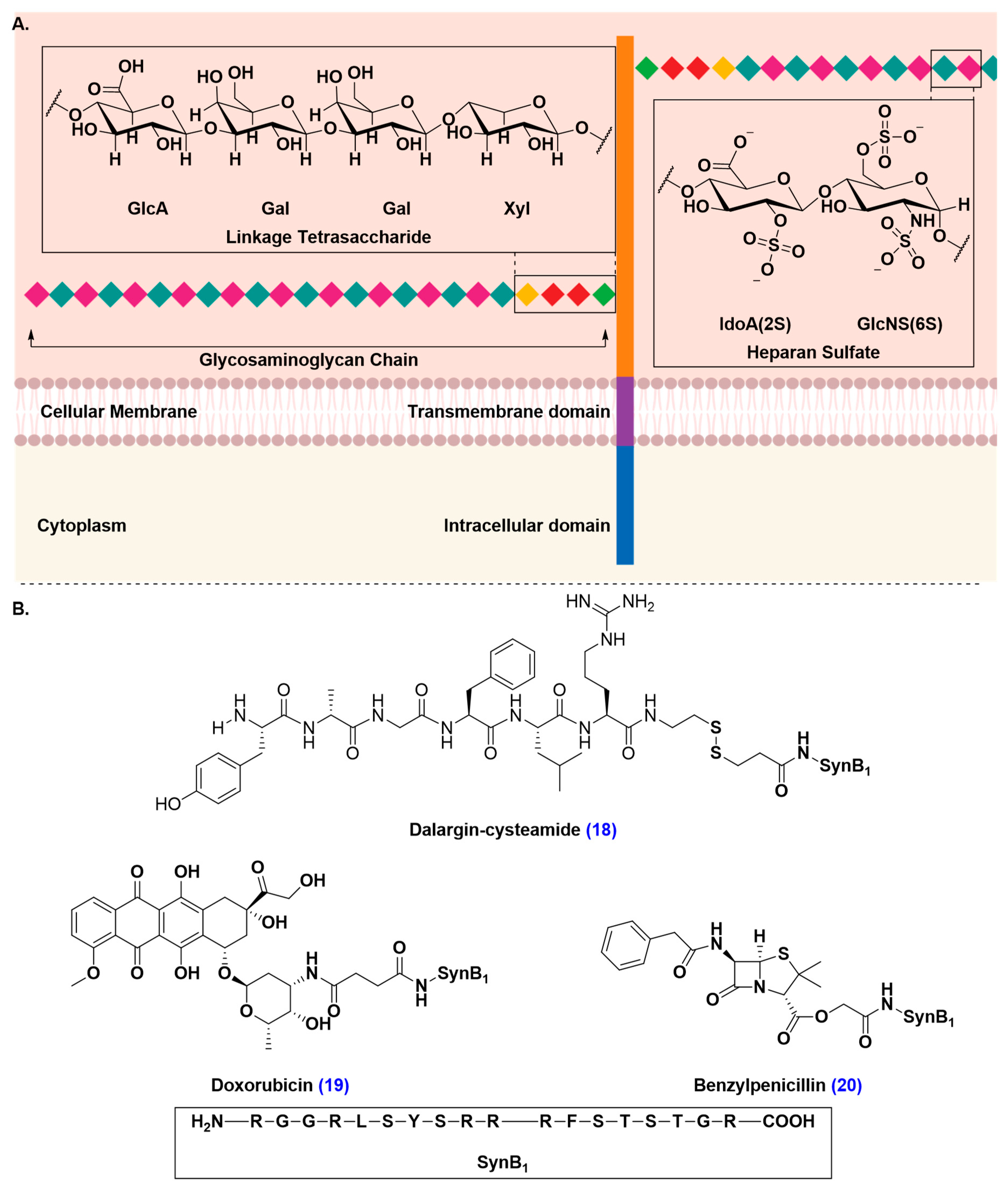
- Montrose, K.; Yang, Y.; Sun, X.; Wiles, S.; Krissansen, G.W. Xentry, a new class of cell-penetrating peptide uniquely equipped for delivery of drugs. Sci. Rep. 2013, 3, 1661–1668. [Google Scholar] [CrossRef]
- Poon, G.M.; Gariépy, J. Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem. Soc. Trans. 2007, 35, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.L.; Ma, J.L.; Wang, T.; Yang, T.B.; Liu, C.B. Cell-penetrating peptide-mediated therapeutic molecule delivery into the central nervous system. Curr. Neuropharmacol. 2013, 11, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Rousselle, C.; Clair, P.; Smirnova, M.; Kolesnikov, Y.; Pasternak, G.W.; Gac-Breton, S.; Rees, A.R.; Scherrmann, J.M.; Temsamani, J. Improved brain uptake and pharmacological activity of dalargin using a peptide-vector-mediated strategy. J. Pharmacol. Exp. Ther. 2003, 306, 371–376. [Google Scholar] [CrossRef]
- Rousselle, C.; Clair, P.; Lefauconnier, J.M.; Kaczorek, M.; Scherrmann, J.M.; Temsamani, J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol. Pharmacol. 2000, 57, 679–686. [Google Scholar] [CrossRef]
- Rousselle, C.; Clair, P.; Temsamani, J.; Scherrmann, J.M. Improved brain delivery of benzylpenicillin with a peptide-vector-mediated strategy. J. Drug Target. 2002, 10, 309–315. [Google Scholar] [CrossRef]
- Kastin, A.J.; Pan, W. Blood-brain barrier and feeding: Regulatory roles of saturable transport systems for ingestive peptides. Curr. Pharm. Des. 2008, 14, 1615–1619. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, C.; Smirnova, M.; Clair, P.; Lefauconnier, J.M.; Chavanieu, A.; Calas, B.; Scherrmann, J.M.; Temsamani, J. Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: Saturation kinetics and specificity. J. Pharmacol. Exp. Ther. 2001, 296, 124–131. [Google Scholar] [PubMed]
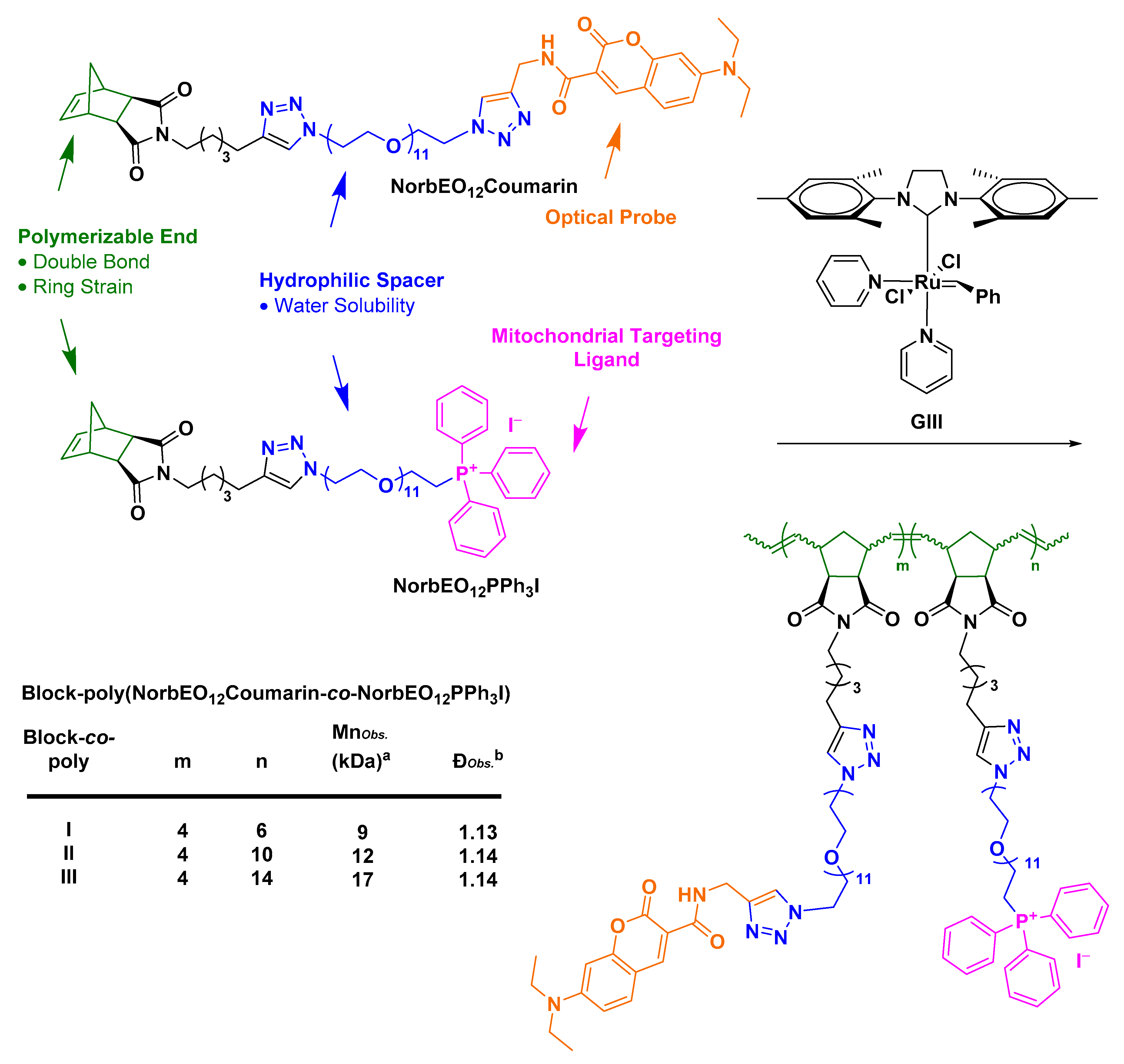
- Abdul Razzak, R. Towards Mitochondrial Targeting for the Treatment of Alzheimer’s Disease; University of St Andrews: St Andrews, UK, 2018. [Google Scholar]
- Garay, R.P.; Labaune, J.P. Immunogenicity of polyethylene glycol (PEG). Open Conf. Proc. J. 2011, 2, 104–107. [Google Scholar] [CrossRef]
- Abuchowski, A.; McCoy, J.R.; Palczuk, N.C.; van Es, T.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar] [PubMed]
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar] [PubMed]
- Pirollo, K.F.; Nemunaitis, J.; Leung, P.K.; Nunan, R.; Adams, J.; Chang, E.H. Safety and efficacy in advanced solid tumors of a targeted nanocomplex carrying the p53 gene used in combination with docetaxel: A phase 1b study. Mol. Ther. 2016, 24, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- (FDA) Doxil® (Doxorubicin Liposome Injection), for Intravenous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/050718s051lbl.pdf (accessed on 12 April 2019).
- (FDA) OnivydeTM (Irinotecan Liposome Injection), for Intravenous Use. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=BasicSearch.process (accessed on 12 April 2019).
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links A-beta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292–320. [Google Scholar] [CrossRef] [PubMed]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, 313–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, W.; Kuang, X.; Hou, S.; Liu, H. Nanopreparations for mitochondria targeting drug delivery system: Current strategies and future prospective. Asian J. Pharm. Sci. 2017, 12, 498–508. [Google Scholar] [CrossRef]
- Sakhrani, N.M.; Padh, H. Organelle targeting: Third level of drug targeting. Drug Des. Dev. Ther. 2013, 7, 585–599. [Google Scholar]
- Bielawski, C.W.; Grubbs, R.H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. [Google Scholar] [CrossRef]
- Slugov, C. Synthesis of Homopolymers and Copolymers. In Handbook of Metathesis; Grubbs, R.H., Khosravi, E., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 1–24. [Google Scholar]
- Asin-Cayuela, J.; Manas, A.-R.B.; James, A.M.; Smith, R.A.J.; Murphy, M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004, 571, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Le Trionnaire, S.; Perry, A.; Szczesny, B.; Szabo, C.; Winyard, P.G.; Whatmore, J.L.; Wood, M.E.; Whiteman, M. The synthesis and functional evaluation of a mitochondria-targeted hydrogen sulfide donor, (10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)decyl)triphenylphosphonium bromide (AP39). Med. Chem. Commun. 2014, 5, 728–736. [Google Scholar] [CrossRef]
- Wawro, A.M.; Muraoka, T.; Kinbara, K. Chromatography-free synthesis of monodisperse oligo(ethylene glycol) mono-p-toluenesulfonates and quantitative analysis of oligomer purity. Polym. Chem. 2016, 7, 2389–2394. [Google Scholar] [CrossRef]
- Fischer, A.; Cremer, C.; Stelzer, E.H.K. Fluorescence of coumarins and xanthenes after two-photon absorption with a pulsed titanium–sapphire laser. Appl. Opt. 1995, 34, 1989–2003. [Google Scholar] [CrossRef]
- Diaspro, A.; Sheppard, C.J.R. Two-photon excitation fluorescence microscopy. In Confocal and Two-Photon Mcroscopy: Foundations, Applications and Advances; Diaspro, A., Ed.; Wiley-Liss Inc.: New York, NY, USA, 2002; pp. 39–73. [Google Scholar]
- Soeller, C.; Cannell, M.B. Two-photon microscopy: Imaging in scattering samples and three-dimensionally resolved flash photolysis. Microsc. Res. Tech. 1999, 47, 182–195. [Google Scholar] [CrossRef]
- Girkin, J.M.; Wokosin, D.L. Practical multiphoton microscopy. In Confocal and Two-Photon Microscopy: Foundations, Applications and Advances; Diaspro, A., Ed.; Wiley-Liss Inc.: New York, NY, USA, 2002; pp. 207–235. [Google Scholar]
- König, K.; Tirlapur, U.K. Cellular and subcellular perturbations during multiphoton microscopy. In Confocal and Two-Photon Microscopy: Foundations, Applications and Advances; Diaspro, A., Ed.; Wiley-Liss Inc.: New York, NY, USA, 2002; pp. 191–205. [Google Scholar]
- Friedman, L.M.; Furberg, C.D.; DeMets, D.L. Introduction to clinical trials. In Fundamentals of Clinical Trials; Springer: New York, NY, USA, 2010; pp. 1–14. [Google Scholar]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Blood Brain Barrier Opening in Alzheimer’ Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03119961?cond=blood+brain+barrier&rank=19 (accessed on 24 April 2019).
- Heinrich Elinzano, M.D. BrUOG 329 GBM Onyvide with TMZ (329). Available online: https://clinicaltrials.gov/ct2/show/NCT03119064 (accessed on 24 April 2019).
- NU-0129 in Treating Patients with Recurrent Glioblastoma or Gliosarcoma Undergoing Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT03020017 (accessed on 24 April 2019).
- Brain Interstitium Temozolomide Concentration pre and Post Regadenoson Administration. Available online: https://clinicaltrials.gov/ct2/show/NCT02389738 (accessed on 24 April 2019).
- Using MRI-Guided Laser Heat Ablation to Induce Disruption of the Peritumoral Blood Brain Barrier to Enhance Delivery and Efficacy of Treatment of Pediatric Brain Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02372409 (accessed on 24 April 2019).
- Gauvain, K.; Tran, D.; Rubin, J.; Shimony, J.; Campian, J.; Leuthardt, E.; Limbrick, D. A pilot study of using MRI-guided laser heat ablation to induce disruption of the peritumoral blood brain barrier to enhance delivery and efficacy of treatment of pediatric brain tumors. Neuro-Oncol. 2016, 18, 129–130. [Google Scholar] [CrossRef][Green Version]
- MK-3475 in Combination with MRI-Guided Laser Ablation in Recurrent Malignant Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT02311582 (accessed on 24 April 2019).
- Campian, J.; Ghiaseddin, A.; Rahman, M.; Ansstas, G.; Kim, A.; Leuthardt, E.; Tran, D. Early results of a multicenter phase I and open-label, randomized phase II study testing the toxicities and efficacy of MK-3475 (pembrolizumab) in combination with mri-guided laser interstitial thermal therapy (litt) in recurrent malignant gliomas. Neuro-Oncol. 2017, 19, 29–30. [Google Scholar] [CrossRef]
- Phase II Etirinotecan Pegol in Refractory Brain Metastases & Advanced Lung Cancer/Metastatic Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02312622 (accessed on 24 April 2019).
- Nagpal, S.; Wakelee, H.; Padda, S.; Bertrand, S.; Acevedo, B.; Tisch, A.H.; Pagtama, J.; Soltys, S.; Neal, J. A Phase II study of etirinotecan pegol (NKTR-102) in patients with refractory brain metastases and advanced lung cancer. J. Thorac. Oncol. 2017, 12, 940. [Google Scholar] [CrossRef][Green Version]
- Effect of Deep TMS on the Permeability of the BBB in Patients with Glioblastoma Multiforme: A Pilot Study. Available online: https://clinicaltrials.gov/ct2/show/NCT02474966 (accessed on 24 April 2019).
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-mediated blood–brain barrier opening: Implications for neuroprotection and drug delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef] [PubMed]
- Phase II Study of Combined Temozolomide and SGT-53 for Treatment of Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02340156 (accessed on 24 April 2019).
- MRI-Guided Laser Surgery and Doxorubicin Hydrochloride in Treating Patients with Recurrent Glioblastoma Multiforme. Available online: https://clinicaltrials.gov/ct2/show/NCT01851733 (accessed on 24 April 2019).
- Safinamide in Idiopathic Parkinson’s Disease (IPD) with Motor Fluctuations, as Add-On to Levodopa (SETTLE). Available online: https://clinicaltrials.gov/ct2/show/NCT00627640 (accessed on 24 April 2019).
- Borgohain, R.; Szasz, J.; Stanzione, P.; Meshram, C.; Bhatt, M.; Chirilineau, D.; Stocchi, F.; Lucini, V.; Giuliani, R.; Forrest, E.; et al. Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov. Disord. 2014, 29, 229–237. [Google Scholar] [CrossRef] [PubMed]
- A Safety Study of Rituximab Plus MTX Injected into the Cerebrospinal Fluid in the Treatment of Brain Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00221325 (accessed on 24 April 2019).
- Rubenstein, J.L.; Li, J.; Chen, L.; Advani, R.; Drappatz, J.; Gerstner, E.; Batchelor, T.; Krouwer, H.; Hwang, J.; Auerback, G.; et al. Multicenter phase 1 trial of intraventricular immunochemotherapy in recurrent CNS lymphoma. Blood 2013, 121, 745–751. [Google Scholar] [CrossRef] [PubMed]
- A Phase 1, Open-Label, Dose Escalation Study of ANG1005 in Patients with Malignant Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT00539344 (accessed on 24 April 2019).
- Drappatz, J.; Brenner, A.; Wong, E.T.; Eichler, A.; Schiff, D.; Groves, M.D.; Mikkelsen, T.; Rosenfeld, S.; Sarantopoulos, J.; Meyers, C.A.; et al. Phase I study of GRN1005 in recurrent malignant glioma. Clin. Cancer Res. 2013, 19, 1567–1576. [Google Scholar] [CrossRef]
- ANG1005 in Breast Cancer Patients with Recurrent Brain Metastases. Available online: https://clinicaltrials.gov/ct2/show/NCT02048059 (accessed on 24 April 2019).
- Li, F.; Tang, S.C. Targeting metastatic breast cancer with ANG1005, a novel peptide-paclitaxel conjugate that crosses the blood-brain-barrier (BBB). Genes Dis. 2017, 4, 1–3. [Google Scholar] [CrossRef]
- Kumthekar, P.; Tang, S.; Brenner, A.J.; Kesari, S.; Anders, C.K.; Carrillo, J.A.; Chalasani, P.; Kabos, P.; Ahluwalia, M.S.; Ibrahim, N.K. A phase II study of ANG1005, a novel BBB/BCB penetratant taxane in patients with recurrent brain metastases and leptomeningeal carcinomatosis from breast cancer. Neuro-Oncol. 2016, 18, 16. [Google Scholar] [CrossRef][Green Version]
- Carboplatin, Melphalan, Etoposide Phosphate, Mannitol, and Sodium Thiosulfate in Treating Patients with Previously Treated Brain Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT00303849 (accessed on 24 April 2019).
- Guillaume, D.J.; Doolittle, N.D.; Gahramanov, S.; Hedrick, N.A.; Delashaw, J.B.; Neuwelt, E.A. Intra-arterial chemotherapy with osmotic blood-brain barrier disruption for aggressive oligodendroglial tumors: Results of a phase I study. Neurosurgery 2010, 66, 48–58. [Google Scholar] [PubMed]
- Doolittle, N.D.; Muldoon, L.L.; Culp, A.Y.; Neuwelt, E.A. Delivery of chemotherapeutics across the blood-brain barrier: Challenges and advances. Adv. Pharmacol. 2014, 71, 203–243. [Google Scholar] [PubMed]
- Methotrexate, Mannitol, Rituximab, and Carboplatin in Treating Patients with Newly Diagnosed Primary Central Nervous System Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00293475 (accessed on 24 April 2019).
- Pegylated Liposomal Doxorubicine and Prolonged Temozolomide in Addition to Radiotherapy in Newly Diagnosed Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00944801 (accessed on 24 April 2019).
- Beier, C.P.; Schmid, C.; Gorlia, T.; Kleinletzenberger, C.; Beier, D.; Grauer, O.; Steinbrecher, A.; Hirschmann, B.; Brawanski, A.; Dietmaier, C.; et al. RNOP-09: pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma--a phase II study. BMC Cancer 2009, 9–19, 308. [Google Scholar] [CrossRef] [PubMed]
- Melphalan with BBBD in Treating Patients with Brain Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT00253721 (accessed on 24 April 2019).
- Couch, J.A.; Yu, Y.J.; Zhang, Y.; Tarrant, J.M.; Fuji, R.N.; Meilandt, W.J.; Solanoy, H.; Tong, R.K.; Hoyte, K.; Luk, W.; et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 2013, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rishi, G.; Subramaniam, V. The relationship between systemic iron homeostasis and erythropoiesis. Biosci. Rep. 2017, 37, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wertheimer, E.; Sasson, S.; Cerasi, E.; Ben-Neriah, Y. The ubiquitous glucose transporter GLUT-1 belongs to the glucose-regulated protein family of stress-inducible proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 2525–2529. [Google Scholar] [CrossRef]
- Wood, I.S.; Trayhurn, P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br. J. Nutr. 2003, 89, 3–9. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin degradation: Progress and potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar]
- Folli, F.; Bonfanti, L.; Renard, E.; Kahn, C.R.; Merighi, A. Insulin receptor substrate-1 (IRS-1) distribution in the rat central nervous system. J. Neurosci. 1994, 14, 6412–6422. [Google Scholar] [CrossRef]
- Kido, Y.; Burks, D.J.; Withers, D.; Bruning, J.C.; Kahn, C.R.; White, M.F.; Accili, D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Investig. 2000, 105, 199–205. [Google Scholar] [CrossRef]
- Watanabe, M.; Hirose, Y.; Sugimoto, M.; Nakanishi, M.; Watanabe, H.; Shimada, M. The distribution of tissue insulin receptors in the mouse by whole-body autoradiography. J. Recept. Res. 1992, 12, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur. J. Clin. Investig. 2003, 33, 1–5. [Google Scholar] [CrossRef]
- Kanai, Y.; Segawa, H.; Miyamoto, K.; Uchino, H.; Takeda, E.; Endou, H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The human SLC7A5 (LAT1): The intriguing histidine/large neutral amino acid transporter and its relevance to human health. Front. Chem. 2018, 6, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Gatter, K.C.; Brown, G.; Trowbridge, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar] [CrossRef] [PubMed]
- West, A.P., Jr.; Bennett, M.J.; Sellers, V.M.; Andrews, N.C.; Enns, C.A.; Bjorkman, P.J. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem. 2000, 275, 38135–38138. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Capobianco, A.; Calì, A.; Bellora, F.; Alberti, F.; Righi, L.; Sapino, A.; Camaschella, C.; Malavasi, F. Structural, functional, and tissue distribution analysis of human transferrin receptor-2 by murine monoclonal antibodies and a polyclonal antiserum. Blood 2002, 100, 3782–3789. [Google Scholar] [CrossRef] [PubMed]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. The low density lipoprotein receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93–105. [Google Scholar] [CrossRef]
- Couchman, J.R.; Ljubimov, A.V. Mammalian tissue distribution of a large heparan sulfate proteoglycan detected by monoclonal antibodies. Matrix 1989, 9, 311–321. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. Restricted transport of anti-transferrin receptor antibody (OX26) through the blood-brain barrier in the rat. J. Neurochem. 2001, 79, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Benson, R.H. Limitations of tritium measurements by liquid scintillation counting of emulsions. Anal. Chem. 1966, 38, 1353–1356. [Google Scholar] [CrossRef]
- Waterfield, W.R.; Spanner, J.A.; Stanford, F.G. Tritium exchange from compounds in dilute aqueous solutions. Nature 1968, 218, 472–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Pollack, G.M. Development of a capillary zone electrophoresis assay to examine the disposition of [D-pen2,5]enkephalin in rats. J. Chromatogr. B Biomed. Appl. 1996, 681, 363–373. [Google Scholar] [CrossRef]
- Penner, N.; Xu, L.; Prakash, C. Radiolabeled absorption, distribution, metabolism, and excretion studies in drug development: Why, when, and how? Chem. Res. Toxicol. 2012, 25, 513–531. [Google Scholar] [CrossRef]
- Cole, G.B.; Satyamurthy, N.; Liu, J.; Wong, K.P.; Small, G.W.; Huang, S.C.; Košmrlj, J.; Barrio, J.R.; Petrič, A. The value of in vitro binding as predictor of in vivo results: A case for [18F]FDDNP PET. Mol. Imaging Biol. 2019, 21, 25–34. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Ito, K.; Uchida, Y.; Ohtsuki, S.; Aizawa, S.; Kawakami, H.; Katsukura, Y.; Kamiie, J.; Terasaki, T. Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J. Pharm. Sci. 2011, 100, 3939–3950. [Google Scholar] [CrossRef] [PubMed]
- Syvänen, S.; Lindhe, O.; Palner, M.; Kornum, B.R.; Rahman, O.; Långström, B.; Knudsen, G.M.; Hammarlund-Udenaes, M. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab. Dispos. 2009, 37, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Pa, J.; Sweeney, M.D.; Toga, A.W.; Zlokovic, B.V. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 2016, 131, 687–707. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Wong, S.M.; Hofman, P.A.M.; Burgmans, S.; Verhey, F.R.J.; Backes, W.H. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol. Aging 2016, 45, 190–196. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Jansen, J.F.A.; Jeukens, C.R.L.P.N.; Burgmans, S.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; van Osch, M.J.P.; Backes, W.H. Subtle blood-brain barrier leakage rate and spatial extent: Considerations for dynamic contrast-enhanced MRI. Med. Phys. 2017, 44, 4112–4125. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S. MRI Assessment of Neurovascular Changes in Idiopathic Parkinson’s Disease. Ph.D. Thesis, University of Manchester, Manchester, UK, 2016. [Google Scholar]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagacé, M.; Kuan, W.L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Taheri, S.; Gasparovic, C.; Shah, N.J.; Rosenberg, G.A. Quantitative measurement of blood-brain barrier permeability in human using dynamic contrast-enhanced MRI with fast T1 mapping. Magn. Reson. Med. 2011, 65, 1036–1042. [Google Scholar] [CrossRef]
- Cramer, S.P.; Simonsen, H.; Frederiksen, J.L.; Rostrup, E.; Larsson, H.B. Abnormal blood–brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. Neuroimage Clin. 2014, 4, 182–189. [Google Scholar] [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Koeppe, R.A.; Reiman, E.M.; Foster, N.L.; Weiner, M.W.; Jagust, W.J. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 2011, 32, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Protas, H.D.; Chen, K.; Langbaum, J.B.; Fleisher, A.S.; Alexander, G.E.; Lee, W.; Bandy, D.; de Leon, M.J.; Mosconi, L.; Buckley, S.; et al. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol. 2013, 70, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Bailly, M.; Destrieux, C.; Hommet, C.; Mondon, K.; Cottier, J.P.; Beaufils, E.; Vierron, E.; Vercouillie, J.; Ibazizene, M.; Voisin, T.; et al. Precuneus and cingulate cortex atrophy and hypometabolism in patients with Alzheimer’s disease and mild cognitive impairment: MRI and (18)F-FDG PET quantitative analysis using freesurfer. Biomed. Res. Int. 2015, 2015, 583931–583939. [Google Scholar] [CrossRef]
- Van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135, 181–189. [Google Scholar] [CrossRef]
- Deo, A.K.; Borson, S.; Link, J.M.; Domino, K.; Eary, J.F.; Ke, B.; Richards, T.L.; Mankoff, D.A.; Minoshima, S.; O’Sullivan, F.; et al. Activity of P-glycoprotein, a β-amyloid transporter at the blood-brain barrier, is compromised in patients with mild Alzheimer disease. J. Nucl. Med. 2014, 55, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, K.; Gunter, J.L.; Tosakulwong, N.; Weigand, S.D.; Senjem, M.S.; Petersen, R.C.; Aisen, P.S.; Jagust, W.J.; Weiner, M.W.; Jack, C.R., Jr.; et al. Focal hemosiderin deposits and β-amyloid load in the ADNI cohort. Alzheimers Dement. 2013, 9, S116–S123. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Michels, L.; Warnock, G.; Buck, A.; Macauda, G.; Leh, S.E.; Kaelin, A.M.; Riese, F.; Meyer, R.; O’Gorman, R.; Hock, C.; et al. Arterial spin labeling imaging reveals widespread and Aβ-independent reductions in cerebral blood flow in elderly apolipoprotein epsilon-4 carriers. J. Cereb. Blood Flow Metab. 2016, 36, 581–595. [Google Scholar] [CrossRef]
- Johnson, N.A.; Jahng, G.H.; Weiner, M.W.; Miller, B.L.; Chui, H.C.; Jagust, W.J.; Gorno-Tempini, M.L.; Schuff, N. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: Initial experience. Radiology 2005, 234, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Melzer, T.R.; Watts, R.; MacAskill, M.R.; Pearson, J.F.; Rüeger, S.; Pitcher, T.L.; Livingston, L.; Graham, C.; Keenan, R.; Shankaranarayanan, A.; et al. Arterial spin labelling reveals an abnormal cerebral perfusion pattern in Parkinson’s disease. Brain 2011, 134, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Salat, D.H.; Rosas, H.D. Complex relationships between cerebral blood flow and brain atrophy in early Huntington’s disease. Neuroimage 2012, 59, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Ingrisch, M.; Sourbron, S.; Morhard, D.; Ertl-Wagner, B.; Kümpfel, T.; Hohlfeld, R.; Reiser, M.; Glaser, C. Quantification of perfusion and permeability in multiple sclerosis: Dynamic contrast-enhanced MRI in 3D at 3T. Investig. Radiol. 2012, 47, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Hojjat, S.P.; Kincal, M.; Vitorino, R.; Cantrell, C.G.; Feinstein, A.; Zhang, L.; Lee, L.; O’Connor, P.; Carroll, T.J.; Aviv, R.I. Cortical Perfusion Alteration in Normal-Appearing Gray Matter Is Most Sensitive to Disease Progression in Relapsing-Remitting Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2016, 37, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.J.; Grace, G.M.; Tartaglia, M.C.; Orange, J.B.; Chen, X.; Rowe, A.; Findlater, K.; Kozak, R.I.; Freedman, M.; Lee, T.Y.; et al. Widespread cerebral haemodynamics disturbances occur early in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rule, R.R.; Schuff, N.; Miller, R.G.; Weiner, M.W. Gray matter perfusion correlates with disease severity in ALS. Neurology 2010, 74, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Hultman, K.; Strickland, S.; Norris, E.H. The APOE ɛ4/ɛ4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2013, 33, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood–brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Omalu, B.I.; DeKosky, S.T.; Minster, R.L.; Kamboh, M.I.; Hamilton, R.L.; Wecht, C.H. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005, 57, 128–134. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, B.; Sajdel-Sulkowska, E.M. New insight into Alzheimer disease: Demonstration of fibrin(ogen)-serum albumin insoluble deposits in brain tissue. Alzheimer Dis. Assoc. Disord. 2006, 20, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. A leaky blood–brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.M.; Kócsi, Z.; Stone, J. Pericapillary haem-rich deposits: Evidence for microhaemorrhages in aging human cerebral cortex. J. Cereb. Blood Flow Metab. 2005, 25, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. Decreased mRNA expression of tight junction proteins in lumbar spinal cords of patients with ALS. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Yao, H.; Zhang, W.; Sutliff, R.L.; Buch, S. Tat 101-mediated enhancement of brain pericyte migration involves platelet-derived growth factor subunit B homodimer: Implications for human immunodeficiency virus-associated neurocognitive disorders. J. Neurosci. 2014, 34, 11812–11825. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Heilman, D.; Haorah, J.; Zelivyanskaya, M.; Persidsky, R.; Weber, G.A.; Shimokawa, H.; Kaibuchi, K.; Ikezu, T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood 2006, 107, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Ramirez, S.H.; Sato, S.; Kiyota, T.; Cerny, R.L.; Kaibuchi, K.; Persidsky, Y.; Ikezu, T. Phosphorylation of Claudin-5 and Occludin by Rho Kinase in Brain Endothelial Cells. Am. J. Pathol. 2008, 172, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Aβ accumulation, hypoperfusion, blood–brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2018, 38, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Baloyannis, S.J.; Baloyannis, I.S. The vascular factor in Alzheimer’s disease: A study in Golgi technique and electron microscopy. J. Neurol. Sci. 2012, 322, 117–121. [Google Scholar] [CrossRef]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227. [Google Scholar] [CrossRef]
- Salloway, S.; Gur, T.; Berzin, T.; Tavares, R.; Zipser, B.; Correia, S.; Hovanesian, V.; Fallon, J.; Kuo-Leblanc, V.; Glass, D.; et al. Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J. Neurol. Sci. 2002, 203–204, 183–187. [Google Scholar] [CrossRef]
- Shargel, L.; Wu-Pong, S.; Yu, A.B.C. Pharmacokinetics of oral absorption. In Applied Biopharmaceutics & Pharmacokinetics; McGraw Hill Professional: New York, NY, USA, 2012; pp. 131–151. [Google Scholar]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Purpose | Year a | Phase | Ref. | |
|---|---|---|---|---|
| 1. | Evaluation of delivery of anti-Aβ with the aid of MA-FUS in patients with AD | 2017 | Phase I/II | [149] |
| 2. | Evaluating the safety and preliminary efficacy of Onivyde® (a nanoliposomal irinotecan) administered in combination with temozolomide in the treatment of patients with recurrent glioblastoma | 2017 | Phase I/II | [150] |
| 3. | Evaluating the safety and efficacy of spherical nucleic acid-conjugated gold nanoparticles (NU-0129) at the treatment of patients with recurrent glioblastoma multiforme or gliosarcoma | 2017 | Early Phase I | [151] |
| 4. | Evaluating brain uptake of temozolomide pre and post transient BBB disruption mediated by administration of regadenoson, a vasoactive peptide, in recurrent high-grade glioma patients | 2015 | Early Phase I | [152] |
| 5. | Evaluating brain uptake of chemotherapeutics upon transient BBB disruption mediated by MRI-guided laser ablation in pediatric patients with brain tumors | 2015 | Phase I | [153,154] |
| 6. | Evaluating brain uptake and efficacy of Pembrolizumab, an anticancer MAb, upon transient BBB disruption mediated by MRI-guided laser ablation in patients with recurrent malignant gliomas | 2015 | Phase I/II | [155,156] |
| 7. | Evaluating the efficacy of etirinotecan pegol at CNS disease control in patients with refractory brain metastases and advanced lung cancer or metastatic breast cancer | 2015 | Phase II | [157,158] |
| 8. | Evaluating the effect of deep transcranial magnetic stimulation on the permeability of the BBB in glioblastoma multiform patients | 2014 | Phase II | [159,160] |
| 9. | Evaluating brain uptake and efficacy of SGT-53, a cationic liposome encapsulating a therapeutic genetic material and decorated with anti-HTfR-MAb fragment, at the induction of apoptosis in patients with recurrent glioblastoma when combined with temozolomide | 2014 | Phase II | [161] |
| 10. | Evaluating brain uptake and efficacy of doxorubicin upon transient BBB disruption mediated by MRI-guided laser ablation in patients with recurrent malignant gliomas | 2013 | Phase I | [162] |
| 11. | Evaluating the efficacy of Safinamide in the treatment of patients with PD | 2009 | Phase III | [163,164] |
| 12. | Evaluation of efficacy of rituximab, an anticancer MAb, given together with methotrexate to treat patients with brain lymphoma after its intraventricular administration | 2007 | Phase I | [165,166] |
| 13. | Evaluation of safety, tolerability, pharmacokinetic and BBB permeability of ANG1005 in patients with recurrent or progressive malignant glioma | 2007 | Phase I/II | [167,168,169,170,171] |
| 14. | Dose optimization of melphalan given together with carboplatin and etoposide phosphate to treat patients with anaplastic oligodendroglioma or oligoastrocytoma after transient BBB disruption mediated by administration of mannitol | 2005 | Phase I/II | [172,173,174] |
| 15. | Evaluation of side effects of methotrexate, rituximab, and carboplatin, given together to treat patients with primary CNS lymphoma after transient BBB disruption mediated by administration of mannitol | 2005 | Phase I/II | [175] |
| 16. | Evaluating the effect of combination therapy of Doxil® and temozolomide in addition to radiotherapy in patients diagnosed with glioblastoma | 2002 | Phase I/II | [176,177] |
| 17. | Side effect evaluation and dose optimization of melphalan administered to treat patients with brain malignancies after transient BBB disruption mediated by administration of mannitol | 1998 | Phase I | [178] |
| NS Drug Delivery Techniques | ||
|---|---|---|
| Drug Delivery Beyond The BBB | ||
| 1. Intraparenchymal Delivery | ||
| Pros | • | Delivery of both small and large therapeutic molecules |
| Cons | • | Invasive |
| • | May be associated with infection or inevitable tissue damage | |
| • | Fraught with technical complications | |
| • | Requires inpatient care and overnight hospitalization | |
| • | Not suitable for a treatment regimen that requires repeated and constant exposure to the therapeutic material | |
| 2. Intrathecal and Intraventricular Delivery | ||
| Pros | • | Delivery of both small and large therapeutic molecules |
| • | May require a single operation for surgical implantation of the catheter, which reduces the risk of brain tissue damage | |
| Cons | • | Invasive |
| • | May be associated with infection if the patient’s body rejects the implanted reservoir of catheter | |
| • | Cost-effective for a small population of patients | |
| • | May require inpatient care and overnight hospitalization, depending on the patient’s case | |
| 3. Intranasal Delivery | ||
| Pros | • | Non-invasive |
| • | Suitable for a treatment regimen that requires repeated and constant exposure to the therapeutic material | |
| • | No precedence of tissue damage associated with the technique | |
| • | Ease of application thus greater patience comfort and compliance | |
| • | Practically an outpatient treatment protocol | |
| • | Cost effective | |
| Cons | • | Limited by the small amount of drug that can diffuse through the nasal cavity and reach the transporting nerves (olfactory or trigeminal) compared to the administered amount |
| Drug Delivery Through the BBB | ||
| 1. Modification of Physicochemical Properties | ||
| Pros | • | Exploits the passive diffusion through LBL of ECs, which is a non-invasive and non-saturable drug delivery modality |
| Cons | • | Limited by physicochemical properties that can be altered |
| • | Excludes molecules, which could be of tremendous therapeutic value, with physicochemical properties that do not agree with computationally derived models | |
| 2. Inhibition of Efflux Protein Pumps | ||
| Pros | • | None-invasive |
| • | Can be co-administered with the drug to attenuate the activity of efflux proteins | |
| • | Ease of administration, thus greater patience comfort and compliance | |
| Cons | • | Down-regulation of efflux transporters’ activity may increase brain uptake of potentially toxic material |
| 3. Transient Disruption of BBB | ||
| Pros | • | Reversible |
| • | No-permanent damage reported to date | |
| • | Non-invasive (compared to direct injection methods) | |
| Cons | • | The CNS remains unprotected against blood-borne toxins during the therapeutic timeframe |
| • | May require inpatient care and overnight hospitalization depending on the patient’s case | |
| 3.1. Hyperosmotic Disruption | ||
| Pros | • | Cost effective |
| Cons | • | Non-specific |
| • | Associated with transient cerebral edema | |
| 3.2. MRI-Guided MA-FUS Disruption | ||
| Pros | • | Less invasive |
| • | More specific | |
| Cons | • | Time consuming and expensive due to FUS beam aberration corrections, multiple MRI acquisitions and ultrasound and MRI contrast agents’ injections |
| 4. Transporter-Mediated Transcytosis | ||
| Pros | • | Non-invasive |
| Cons | • | A saturable process |
| • | Nonspecific body distribution due to ubiquitous expression of transporters in several body regions | |
| • | May improve brain uptake but not to the therapeutically and clinically effective concentration of the cargo (subject to further investigation) | |
| 4.1. Solute Carrier-Mediated Transcytosis | ||
| Pros | • | Solute carriers’ expression levels, glucose in particular, are significant relative to receptor proteins, which makes them an ideal target |
| Cons | • | Sensitive to the vector’s stereochemical conformation, rigidity and ligation regiochemistry of its cargo |
| • | Restricted by the cargo’s size which should mimic that of its natural substrates | |
| • | Vector-drug ligation may result in loss of vectors affinity towards the protein carrier | |
| • | Vector-drug conjugates may compete with natural substrates for carrier binding, which may result in serious nutritional deprivation | |
| 4.2. Receptor-Mediated Transcytosis | ||
| Pros | • | Transport is not restricted by the vector’s or cargo’s stereochemical conformation or size; therefore, it is suitable for the transport of small molecules, as well as recombinant proteins |
| • | More diverse with respect to vectors design (e.g., a monoclonal antibody, a fragment of the antibody or a peptide derived from natural substrates) | |
| Cons | • | Limited capacity due to the limited receptor expression levels compared to solute carriers |
| • | May be accompanied with acute clinical signs due to competition with natural substrates, which questions the safety of RMT-conjugates | |
| • | Lysosomal sorting may occur preventing exocytosis of the cargo and potentially degrading the RMT vector and the conjugated therapeutic material | |
| 4.3. Adsorptive-Mediated Transcytosis | ||
| Pros | • | Transport is not restricted by the vector’s or cargo’s stereochemical conformation or size, which makes it suitable for the transport of small molecules, as well as peptides |
| • | Binding sites are saturated at higher concentrations in AMT than in CMT and RMT | |
| • | No precedence of interference with cell signaling pathways | |
| Cons | • | CPP may bind to serum proteins which can mask their positive charge and ultimately reduce their brain uptake |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdul Razzak, R.; Florence, G.J.; Gunn-Moore, F.J. Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. Int. J. Mol. Sci. 2019, 20, 3108. https://doi.org/10.3390/ijms20123108
Abdul Razzak R, Florence GJ, Gunn-Moore FJ. Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. International Journal of Molecular Sciences. 2019; 20(12):3108. https://doi.org/10.3390/ijms20123108
Chicago/Turabian StyleAbdul Razzak, Rana, Gordon J. Florence, and Frank J. Gunn-Moore. 2019. "Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis" International Journal of Molecular Sciences 20, no. 12: 3108. https://doi.org/10.3390/ijms20123108
APA StyleAbdul Razzak, R., Florence, G. J., & Gunn-Moore, F. J. (2019). Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. International Journal of Molecular Sciences, 20(12), 3108. https://doi.org/10.3390/ijms20123108