Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Neurodegenerative Diseases (NDDs)
2.1. Alzheimer’s Disease
2.1.1. Amyloid-β Cascade Hypothesis
2.1.2. The Microtubule-Associated Protein Tau Hypothesis
2.2. Parkinson’s Disease
2.3. Other Known NDDs
3. Metal-Based Drugs for NDDs Treatment
3.1. Lithium-Based Treatment
3.2. Inert Complexes Metal Ions
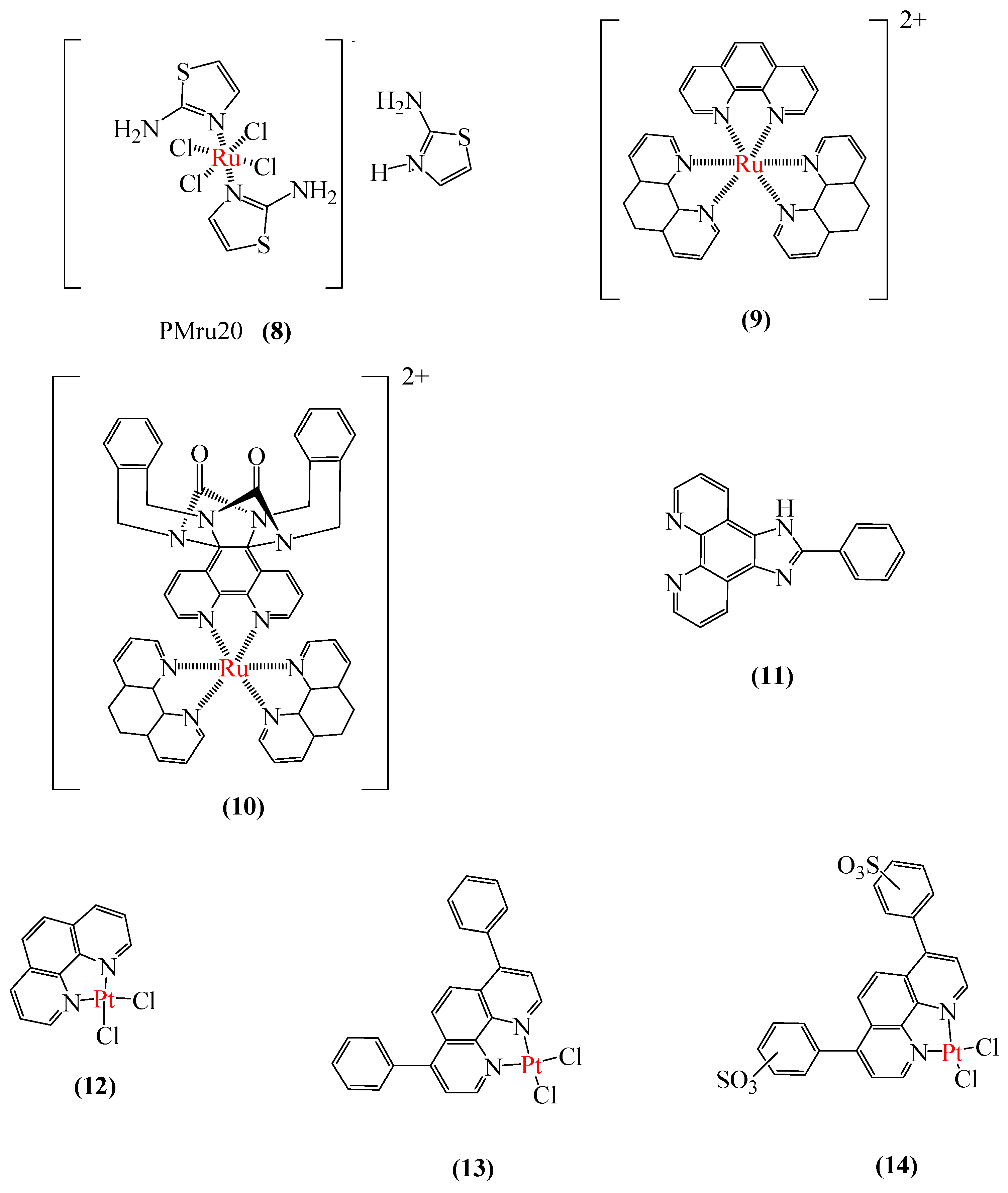
3.2.1. SODs Mimic Metal-Containing Drugs
3.2.2. Metal-Based Prion Protein Aggregation Inhibitors
3.2.3. Aβ Aggregation Inhibitors
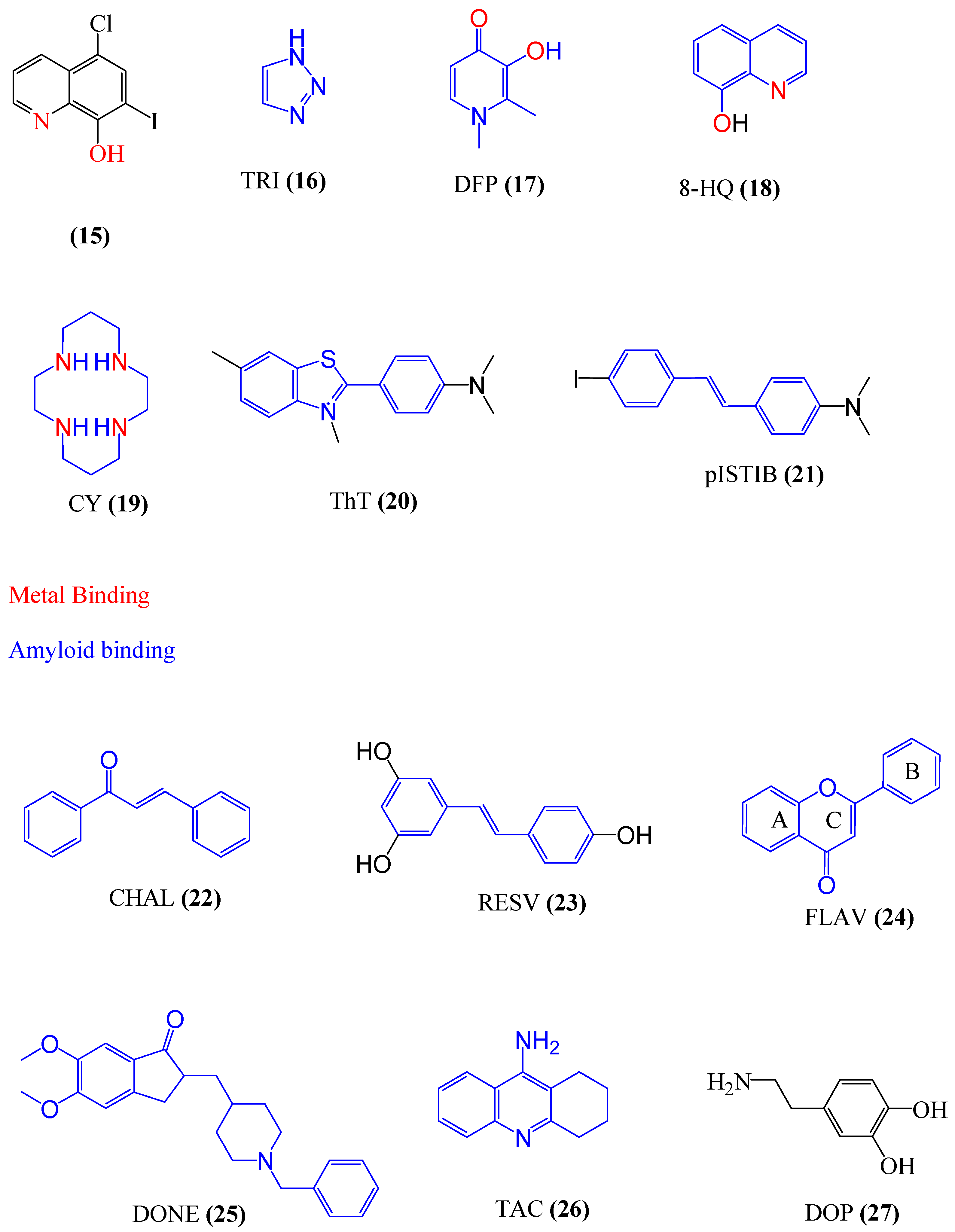
4. Chelating Agents
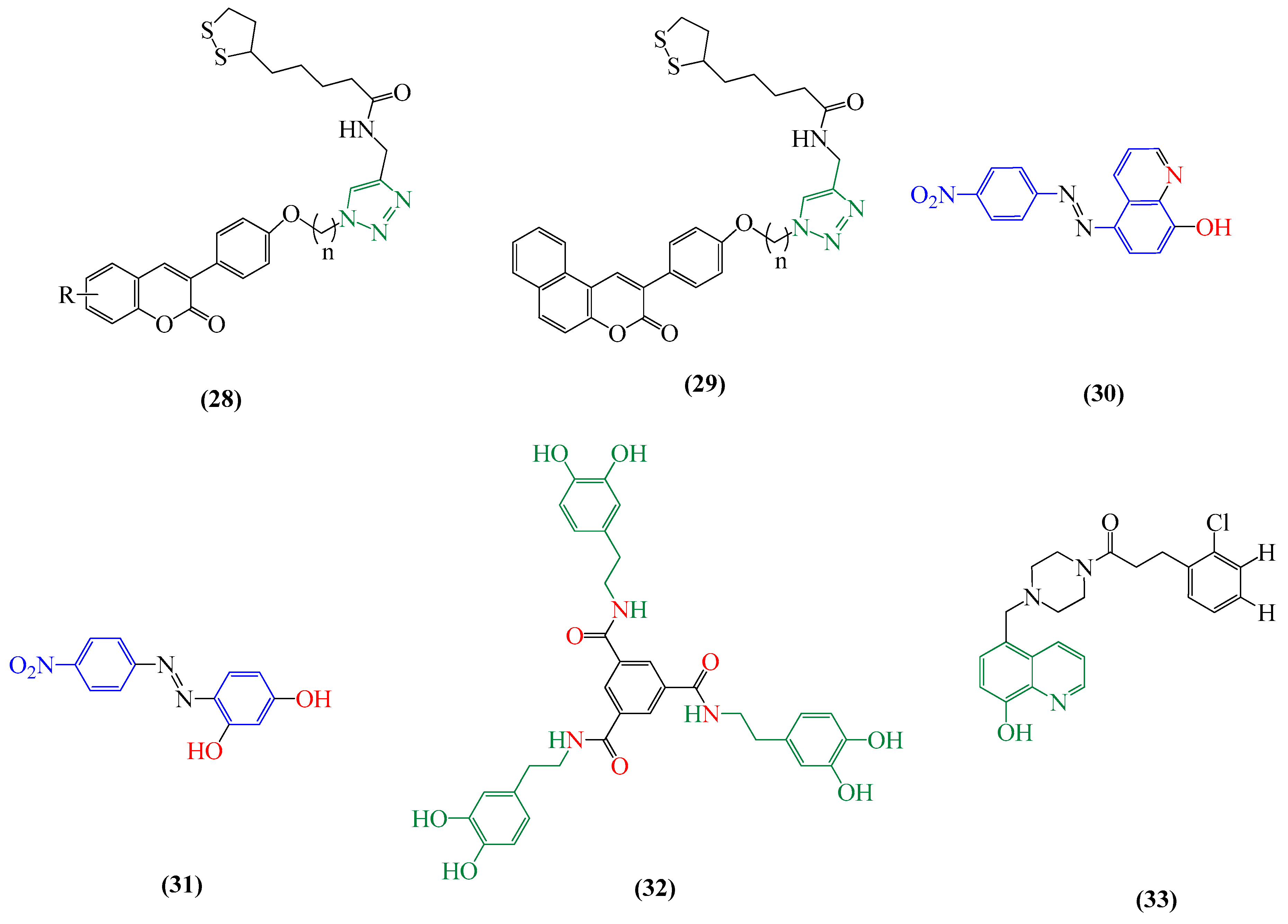
4.1. Multifunctional Agents
4.2. Drug Repositioning for Chelating Agents
4.3. Metal Protein Attenuating Compounds (MPACs)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 8-HQ | 8-Hydroxyquinoline |
| Aß | Amyloid-ß |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| APP | Amyloid precursor protein |
| BBB | Blood-brain barrier |
| CHAL | Chalcone |
| ChEs | Cholinesterases |
| CNS | Central nervous system |
| CQ | Clioquinol |
| CSF | Cerebrospinal fluid |
| CY | Cyclam |
| DFP | Diferiprone |
| DLB | Dementia with Lewy bodies |
| DONE | Donepezil |
| DOP | Dopamine |
| FLAV | Flavone |
| FNHK | |
| FRDA | Friedreich’s ataxia |
| FTD | Frontotemporal dementia |
| GSK-3 | Synthase kinase-3 |
| HD | Huntington’s disease |
| IMP | Inositol monophosphatase |
| IP3 | Inositol triphosphate |
| MAOs | Monoamine Oxidases |
| Mb | Methanobactin |
| MFCs | Multifunctional compounds |
| MPACs | Metal Protein Attenuating Compounds |
| MTDLs | Multitarget-directed ligands |
| NDDs | Neurodegenerative diseases |
| NFTs | Neurofibrillary tangles |
| PD | Parkinson’s disease |
| PDB | Protein Data Bank |
| pISTIB | p-I-stilbene |
| P-Tau | Hyperphosphorylated tau |
| RESV | Resveratrol |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutases |
| TAC | Tacrine |
| ThT | Thioflavine T |
| TRI | Triazole |
| UHK |
References
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 301–307. [Google Scholar]
- Gabor, G. Kovacs current concepts of neurodegenerative diseases. Eur. Med. J. Neurol. 2014, 1, 78–86. [Google Scholar]
- Kovacs, G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Modi, G.; Pillay, V.; Choonara, Y.E. Advances in the treatment of neurodegenerative disorders employing nanotechnology. Ann. N. Y. Acad. Sci. 2010, 1184, 154–172. [Google Scholar] [CrossRef]
- Subramaniam, N.S.; Bawden, C.S.; Waldvogel, H.; Faull, R.M.L.; Howarth, G.S.; Snell, R.G. Emergence of breath testing as a new non-invasive diagnostic modality for neurodegenerative diseases. Brain Res. 2018, 1691, 75–86. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef]
- Ward, K.R.; Yealy, D.M. End-tidal carbon dioxide monitoring in emergency medicine, Part 2: Clinical applications. Acad. Emerg. Med. 1998, 5, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Helder, D.I.; Kaptein, A.A.; Van Kempen, G.M.J.; Weinman, J.; Van Houwelingen, J.C.; Roos, R.A.C. Living with Huntington’s disease: illness perceptions, coping mechanisms, and spouses’ quality of life. Int. J. Behav. Med. 2002, 9, 37–52. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [CrossRef]
- Jiang, Z.; You, Q.; Zhang, X. Medicinal chemistry of metal chelating fragments in metalloenzyme active sites: A perspective. Eur. J. Med. Chem. 2019, 165, 172–197. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling Amyloid-β, Tau, and Metals in Alzheimer’s Disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst. Rev. 2012, 16, CD005380, Erratum in 2014, 21, CD005380. [Google Scholar]
- Hiremathad, A.; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel Tacrine-Hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 148, 255–267. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42 Peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef] [PubMed]
- Martorell, M.; Forman, K.; Castro, N.; Capó, X.; Tejada, S.; Sureda, A. Potential Therapeutic Effects of Oleuropein Aglycone in Alzheimer’s Disease. Curr. Pharm. Biotechnol. 2016, 17, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Nardi, M.; Bonacci, S.; De Luca, G.; Maiuolo, J.; Oliverio, M.; Sindona, G.; Procopio, A. Biomimetic synthesis and antioxidant evaluation of 3,4-DHPEA-EDA [2-(3,4-hydroxyphenyl) ethyl (3S,4E)-4-formyl-3-(2-oxoethyl)hex-4-enoate]. Food Chem. 2014, 162, 89–93. [Google Scholar] [CrossRef]
- Sarbishegi, M. Antioxidant Effects of Olive Leaf Extract in Prevention of Alzheimer’s Disease and Parkinson’s Disease. Gene Cell Tissue 2018, 5, e79847. [Google Scholar] [CrossRef]
- Sindona, G.; Caruso, A.; Cozza, A.; Fiorentini, S.; Lorusso, B.; Marini, E.; Nardi, M.; Procopio, A.; Zicari, S. Anti-Inflammatory Effect of 3,4-DHPEA-EDA [2-(3,4 -Hydroxyphenyl) ethyl (3S, 4E)- 4-Formyl-3-(2-Oxoethyl)Hex-4-Enoate] on Primary Human Vascular Endothelial Cells. Curr. Med. Chem. 2012, 19, 4006–4013. [Google Scholar] [CrossRef]
- Nardi, M.; Bonacci, S.; Cariati, L.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Synthesis and antioxidant evaluation of lipophilic oleuropein aglycone derivatives. Food Funct. 2017, 8, 4684–4692. [Google Scholar] [CrossRef] [PubMed]
- Paonessa, R.; NArdi, M.; Di gioia, M.L.; Olivito, F.; Oliverio, M.; Procopio, A. Eco-friendly synthesis of lipophilic EGCG derivatives and antitumor and antioxidant evaluation. Nat. Prod. Comun. 2018, 13, 1097–1234. [Google Scholar] [CrossRef]
- Chen, K.; Cui, M. Recent progress in the development of metal complexes as β-amyloid imaging probes in the brain. Medchemcomm 2017, 8, 1393–1407. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76 Pt A, 27–50. [Google Scholar] [CrossRef]
- de Castro, A.A.; da Cunha, E.F.F.; Pereira, A.F.; Soares, F.V; Leal, D.H.S.; Kuca, K.; Ramalho, T.C. Insights into the Drug Repositioning Applied to the Alzheimer’s Disease Treatment and Future Perspectives. Curr. Alzheimer Res. 2018, 15, 1161–1178. [Google Scholar] [CrossRef]
- Di Stefano, A.; Iannitelli, A.; Laserra, S.; Sozio, P. Drug delivery strategies for Alzheimer’s disease treatment. Expert Opin. Drug Deliv. 2011, 8, 581–603. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hanninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Kukharsky, M.S.; Ovchinnikov, R.K.; Bachurin, S.O. [Molecular aspects of the pathogenesis and current approaches to pharmacological correction of Alzheimer’s disease]. Zhurnal Nevrol. i psikhiatrii Im. S.S. Korsakova 2015, 115, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.M.; Seward, M.E.; Bloom, G.S. Alzheimer disease: a tale of two prions. Prion 2013, 7, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O. V Alzheimer’s disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar] [PubMed]
- Bachurin, S.O.; Bovina, E.V; Ustyugov, A.A. Drugs in Clinical Trials for Alzheimer’s Disease: The Major Trends. Med. Res. Rev. 2017, 37, 1186–1225. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Ballard, C.; Corbett, A.; Sharp, S. Aligning the evidence with practice: NICE guidelines for drug treatment of Alzheimer’s disease. Expert Rev. Neurother. 2011, 11, 327–329. [Google Scholar] [CrossRef]
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.B.; Edison, P.; Hagan, J.J.; Holmes, C.; Jones, E.; Katona, C.; et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet. Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.L.; Hanney, M.; Francis, P.T.; Ballard, C.G. Amyloid beta concentrations in older people with Down syndrome and dementia. Neurosci. Lett. 2009, 451, 162–164. [Google Scholar] [CrossRef]
- Broadstock, M.; Ballard, C.; Corbett, A. Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Expert Opin. Pharmacother. 2014, 15, 1797–1810. [Google Scholar] [CrossRef]
- Saez-Orellana, F.; Godoy, P.A.; Bastidas, C.Y.; Silva-Grecchi, T.; Guzman, L.; Aguayo, L.G.; Fuentealba, J. ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of beta-amyloid peptide in hippocampal neurons. Neuropharmacology 2016, 100, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Godyn, J.; Jonczyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E. The pathogenesis of Alzheimer’s disease and the role of Abeta42. CNS Spectr. 2007, 12, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment strategies targeting amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Abeta(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef]
- Yan, R.; Vassar, R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet. Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef]
- Kuhn, P.-H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 31, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M.; Polanco, J.-C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef]
- Tomita, T. Secretase inhibitors and modulators for Alzheimer’s disease treatment. Expert Rev. Neurother. 2009, 9, 661–679. [Google Scholar] [CrossRef]
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695. [Google Scholar] [CrossRef]
- Corbett, A.; Ballard, C. Is a potential Alzheimer’s therapy already in use for other conditions? Can medications for hypertension, diabetes and acne help with the symptoms? Expert Opin. Investig. Drugs 2013, 22, 941–943. [Google Scholar] [CrossRef][Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M.D. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef]
- Kadavath, H.; Jaremko, M.; Jaremko, L.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Folding of the Tau Protein on Microtubules. Angew. Chem. Int. Ed. Engl. 2015, 54, 10347–10351. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Trojanowski, J.Q. The disordered neuronal cytoskeleton in Alzheimer’s disease. Curr. Opin. Neurobiol. 1992, 2, 653–656. [Google Scholar] [CrossRef]
- Clark, C.M.; Xie, S.; Chittams, J.; Ewbank, D.; Peskind, E.; Galasko, D.; Morris, J.C.; McKeel, D.W.J.; Farlow, M.; Weitlauf, S.L.; et al. Cerebrospinal fluid tau and beta-amyloid: How well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch. Neurol. 2003, 60, 1696–1702. [Google Scholar] [CrossRef]
- Arriagada, P.V; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- de Lau, L.M.L.; Breteler, M.M.B. Epidemiology of Parkinson’s disease. Lancet. Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Svenningsson, P.; Westman, E.; Ballard, C.; Aarsland, D. Cognitive impairment in patients with Parkinson’s disease: Diagnosis, biomarkers, and treatment. Lancet. Neurol. 2012, 11, 697–707. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122 Pt 8, 1437–1448. [Google Scholar] [CrossRef]
- Schneider, S.A.; Obeso, J.A. Clinical and pathological features of Parkinson’s disease. Curr. Top. Behav. Neurosci. 2015, 22, 205–220. [Google Scholar]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Bratzke, H.; Hamm-Clement, J.; Sandmann-Keil, D.; Rub, U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J. Neurol. 2002, 249 (Suppl. III), 1–5. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef]
- Hsu, W.-Y.; Lane, H.-Y.; Lin, C.-H. Medications Used for Cognitive Enhancement in Patients With Schizophrenia, Bipolar Disorder, Alzheimer’s Disease, and Parkinson’s Disease. Front. Psychiatry 2018, 9, 91. [Google Scholar] [CrossRef]
- Lazzara, C.A.; Kim, Y.-H. Potential application of lithium in Parkinson’s and other neurodegenerative diseases. Front. Neurosci. 2015, 9, 403. [Google Scholar] [CrossRef]
- Stelmashook, E.V; Isaev, N.K.; Genrikhs, E.E.; Amelkina, G.A.; Khaspekov, L.G.; Skrebitsky, V.G.; Illarioshkin, S.N. Role of zinc and copper ions in the pathogenetic mechanisms of Alzheimer’s and Parkinson’s diseases. Biochemistry 2014, 79, 391–396. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef]
- Grossman, M. Frontotemporal dementia: A review. J. Int. Neuropsychol. Soc. 2002, 8, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Seelaar, H.; Rohrer, J.D.; Pijnenburg, Y.A.L.; Fox, N.C.; van Swieten, J.C. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: A review. J. Neurol. Neurosurg. Psychiatry 2011, 82, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Metzler-Baddeley, C. A review of cognitive impairments in dementia with Lewy bodies relative to Alzheimer’s disease and Parkinson’s disease with dementia. Cortex 2007, 43, 583–600. [Google Scholar] [CrossRef]
- Rolinski, M.; Fox, C.; Maidment, I.; McShane, R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst. Rev. 2012, CD006504. [Google Scholar] [CrossRef]
- Aranca, T.V; Jones, T.M.; Shaw, J.D.; Staffetti, J.S.; Ashizawa, T.; Kuo, S.-H.; Fogel, B.L.; Wilmot, G.R.; Perlman, S.L.; Onyike, C.U.; et al. Emerging therapies in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2016, 6, 49–65. [Google Scholar] [CrossRef]
- Bürk, K. Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias 2017, 4, 4. [Google Scholar]
- Appleby, B.S.; Connor, A.; Wang, H. Therapeutic strategies for prion disease: A practical perspective. Curr. Opin. Pharmacol. 2019, 44, 15–19. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Verma, A. Prions, prion-like prionoids, and neurodegenerative disorders. Ann. Indian Acad. Neurol. 2016, 19, 169–174. [Google Scholar] [CrossRef]
- Alison, A. The Red-Hot Debate About Transmissible Alzheimer’s. Nature 2016, 531, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, A.; Hider, R.C. The crucial role of metal ions in neurodegeneration: The basis for a promising therapeutic strategy. Br. J. Pharmacol. 2005, 146, 1041–1059. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Metal based neurodegenerative diseases—From molecular mechanisms to therapeutic strategies. Coord. Chem. Rev. 2008, 252, 1189–1199. [Google Scholar] [CrossRef]
- Zhu, Z.-F.; Wang, Q.-G.; Han, B.-J.; William, C.P. Neuroprotective effect and cognitive outcome of chronic lithium on traumatic brain injury in mice. Brain Res. Bull. 2010, 83, 272–277. [Google Scholar] [CrossRef]
- Basselin, M.; Chang, L.; Bell, J.M.; Rapoport, S.I. Chronic lithium chloride administration attenuates brain NMDA receptor-initiated signaling via arachidonic acid in unanesthetized rats. Neuropsychopharmacology 2006, 31, 1659–1674. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, I.M.; Cuningham, J. Persisting neurologic sequelae of lithium carbonate therapy. Arch. Neurol. 1983, 40, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Ewers, M.; Burger, K.; Annas, P.; Mortberg, A.; Bogstedt, A.; Frolich, L.; Schroder, J.; Schonknecht, P.; Riepe, M.W.; et al. Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1668. [Google Scholar] [CrossRef]
- Gotz, J.; Nitsch, R.M. Compartmentalized tau hyperphosphorylation and increased levels of kinases in transgenic mice. Neuroreport 2001, 12, 2007–2016. [Google Scholar]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Liu, S.J.; Zhang, A.H.; Li, H.L.; Wang, Q.; Deng, H.M.; Netzer, W.J.; Xu, H.; Wang, J.Z. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J. Neurochem. 2003, 87, 1333–1344. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef]
- Lovestone, S.; Davis, D.R.; Webster, M.T.; Kaech, S.; Brion, J.P.; Matus, A.; Anderton, B.H. Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations. Biol. Psychiatry 1999, 45, 995–1003. [Google Scholar] [CrossRef]
- Engel, T.; Goni-Oliver, P.; Gomez de Barreda, E.; Lucas, J.J.; Hernandez, F.; Avila, J. Lithium, a potential protective drug in Alzheimer’s disease. Neurodegener. Dis. 2008, 5, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Cade, J.F. Lithium salts in the treatment of psychotic excitement. 1949. Bull. World Health Organ. 2000, 78, 518–520. [Google Scholar] [PubMed]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rubinsztein, D.C. Inositol and IP3 levels regulate autophagy: Biology and therapeutic speculations. Autophagy 2006, 2, 132–134. [Google Scholar] [CrossRef]
- Forlenza, O.V.; de Paula, V.J.; Machado-Vieira, R.; Diniz, B.S.; Gattaz, W.F. Does lithium prevent Alzheimer’s disease? Drugs Aging 2012, 29, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.C.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin. Cell Dev. Biol. 2010, 21, 691–698. [Google Scholar] [CrossRef]
- Pasquali, L.; Busceti, C.L.; Fulceri, F.; Paparelli, A.; Fornai, F. Intracellular pathways underlying the effects of lithium. Behav. Pharmacol. 2010, 21, 473–492. [Google Scholar] [CrossRef]
- Forlenza, O.V; De-Paula, V.J.R.; Diniz, B.S.O. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Birch, N.J. Letter: Lithium and magnesium-dependent enzymes. Lancet 1974, 2, 965–966. [Google Scholar] [CrossRef]
- Amari, L.; Layden, B.; Rong, Q.; Geraldes, C.F.G.C.; Mota de Freitas, D. Comparison of Fluorescence, 31P NMR, and 7Li NMR Spectroscopic Methods for Investigating Li+/Mg2+ Competition for Biomolecules. Anal. Biochem. 1999, 272, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Lee, F.H.F.; Kaidanovich-Beilin, O.; Roder, J.C.; Woodgett, J.R.; Wong, A.H.C. Genetic inactivation of GSK3alpha rescues spine deficits in Disc1-L100P mutant mice. Schizophr. Res. 2011, 129, 74–79. [Google Scholar] [CrossRef]
- Forlenza, O.V; Diniz, B.S.; Radanovic, M.; Santos, F.S.; Talib, L.L.; Gattaz, W.F. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: Randomised controlled trial. Br. J. Psychiatry 2011, 198, 351–356. [Google Scholar] [CrossRef]
- Shalbuyeva, N.; Brustovetsky, T.; Brustovetsky, N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J. Biol. Chem. 2007, 282, 18057–18068. [Google Scholar] [CrossRef]
- Bachmann, R.F.; Wang, Y.; Yuan, P.; Zhou, R.; Li, X.; Alesci, S.; Du, J.; Manji, H.K. Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int. J. Neuropsychopharmacol. 2009, 12, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, J.A.; Machado-Vieira, R.; Zarate, C.A.J.; Manji, H.K. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology 2010, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Ngok-Ngam, P.; Watcharasit, P.; Thiantanawat, A.; Satayavivad, J. Pharmacological inhibition of GSK3 attenuates DNA damage-induced apoptosis via reduction of p53 mitochondrial translocation and Bax oligomerization in neuroblastoma SH-SY5Y cells. Cell. Mol. Biol. Lett. 2013, 18, 58–74. [Google Scholar] [CrossRef]
- Feier, G.; Valvassori, S.S.; Varela, R.B.; Resende, W.R.; Bavaresco, D.V; Morais, M.O.; Scaini, G.; Andersen, M.L.; Streck, E.L.; Quevedo, J. Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacol. Biochem. Behav. 2013, 103, 589–596. [Google Scholar] [CrossRef]
- Engel, T.; Goni-Oliver, P.; Lucas, J.J.; Avila, J.; Hernandez, F. Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 2006, 99, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Ando, K.; Heraud, C.; Yilmaz, Z.; Authelet, M.; Boeynaems, J.-M.; Buee, L.; De Decker, R.; Brion, J.-P. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimers Dis. 2010, 19, 705–719. [Google Scholar] [CrossRef]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef]
- Rockenstein, E.; Torrance, M.; Adame, A.; Mante, M.; Bar-on, P.; Rose, J.B.; Crews, L.; Masliah, E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 2007, 27, 1981–1991. [Google Scholar] [CrossRef]
- Su, Y.; Ryder, J.; Li, B.; Wu, X.; Fox, N.; Solenberg, P.; Brune, K.; Paul, S.; Zhou, Y.; Liu, F.; et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 2004, 43, 6899–6908. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, Y.; Chuang, D.-M. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J. Neurotrauma 2012, 29, 2342–2351. [Google Scholar] [CrossRef]
- Zhang, X.; Heng, X.; Li, T.; Li, L.; Yang, D.; Zhang, X.; Du, Y.; Doody, R.S.; Le, W. Long-term treatment with lithium alleviates memory deficits and reduces amyloid-beta production in an aged Alzheimer’s disease transgenic mouse model. J. Alzheimers Dis. 2011, 24, 739–749. [Google Scholar] [CrossRef]
- Fiorentini, A.; Rosi, M.C.; Grossi, C.; Luccarini, I.; Casamenti, F. Lithium Improves Hippocampal Neurogenesis, Neuropathology and Cognitive Functions in APP Mutant Mice. PLoS ONE 2010, 5, e14382. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.; Munoz-Montano, J.R.; Satrustegui, J.; Avila, J.; Bogonez, E.; Diaz-Nido, J. Regulation of tau phosphorylation and protection against beta-amyloid-induced neurodegeneration by lithium. Possible implications for Alzheimer’s disease. Bipolar Disord. 2002, 4, 153–165. [Google Scholar] [CrossRef]
- Chen, R.W.; Chuang, D.M. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J. Biol. Chem. 1999, 274, 6039–6042. [Google Scholar] [CrossRef]
- Chen, G.; Zeng, W.Z.; Yuan, P.X.; Huang, L.D.; Jiang, Y.M.; Zhao, Z.H.; Manji, H.K. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 1999, 72, 879–882. [Google Scholar] [CrossRef]
- Macdonald, A.; Briggs, K.; Poppe, M.; Higgins, A.; Velayudhan, L.; Lovestone, S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2008, 23, 704–711. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Salvemini, D.; Riley, D.P.; Cuzzocrea, S. Sod mimetics are coming of age. Nat. Rev. Drug Discov. 2002, 1, 367. [Google Scholar] [CrossRef]
- Strange, R.W.; Antonyuk, S.V; Hough, M.A.; Doucette, P.A.; Valentine, J.S.; Hasnain, S.S. Variable Metallation of Human Superoxide Dismutase: Atomic Resolution Crystal Structures of Cu–Zn, Zn–Zn and As-isolated Wild-type Enzymes. J. Mol. Biol. 2006, 356, 1152–1162. [Google Scholar] [CrossRef]
- Wanninger, S.; Lorenz, V.; Subhan, A.; Edelmann, F.T. Metal complexes of curcumin – synthetic strategies, structures and medicinal applications. Chem. Soc. Rev. 2015, 44, 4986–5002. [Google Scholar] [CrossRef]
- Sumanont, Y.; Murakami, Y.; Tohda, M.; Vajragupta, O.; Matsumoto, K.; Watanabe, H. Evaluation of the Nitric Oxide Radical Scavenging Activity of Manganese Complexes of Curcumin and Its Derivative. Biol. Pharm. Bull. 2004, 27, 170–173. [Google Scholar] [CrossRef]
- Vajragupta, O.; Boonchoong, P.; Watanabe, H.; Tohda, M.; Kummasud, N.; Sumanont, Y. Manganese complexes of curcumin and its derivatives: Evaluation for the radical scavenging ability and neuroprotective activity. Free Radic. Biol. Med. 2003, 35, 1632–1644. [Google Scholar] [CrossRef]
- Sumanont, Y.; Murakami, Y.; Tohda, M.; Vajragupta, O.; Watanabe, H.; Matsumoto, K. Effects of Manganese Complexes of Curcumin and Diacetylcurcumin on Kainic Acid-Induced Neurotoxic Responses in the Rat Hippocampus. Biol. Pharm. Bull. 2007, 30, 1732–1739. [Google Scholar] [CrossRef]
- Belda, R.; Blasco, S.; Verdejo, B.; Jiménez, H.R.; Doménech-Carbó, A.; Soriano, C.; Latorre, J.; Terencio, C.; García-España, E. Homo- and heterobinuclear Cu2+ and Zn2+ complexes of abiotic cyclic hexaazapyridinocyclophanes as SOD mimics. Dalt. Trans. 2013, 42, 11194–11204. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, B.; Zhao, C.; Wang, Y.; He, L.; Cui, M.; Zhu, X.; Du, W. Inhibition of human prion neuropeptide PrP106-126 aggregation by hexacoordinated ruthenium complexes. J. Inorg. Biochem. 2013, 128, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zahn, R.; Liu, A.; Lührs, T.; Riek, R.; von Schroetter, C.; López García, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef]
- Messori, L.; Camarri, M.; Ferraro, T.; Gabbiani, C.; Franceschini, D. Promising in Vitro anti-Alzheimer Properties for a Ruthenium(III) Complex. ACS Med. Chem. Lett. 2013, 4, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.E.; Perez, R.G.; Kobayashi, H. Cholinesterase Inhibitor Therapy in Alzheimer’s: The limits and tolerability of Irreversible CNS-selective Acetylcholinesterase Inhibition in Primates. J. Alzheimers Dis. 2017, 55, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.A.; Bhat, S.S.; Kumbhar, A.S.; Sonawane, U.B.; Jani, V.; Joshi, R.R.; Ramteke, S.N.; Kulkarni, P.P.; Joshi, B. Ruthenium(II) polypyridyl complex as inhibitor of acetylcholinesterase and Aβ aggregation. Eur. J. Med. Chem. 2014, 75, 375–381. [Google Scholar] [CrossRef]
- Lu, L.; Zhong, H.-J.; Wang, M.; Ho, S.-L.; Li, H.-W.; Leung, C.-H.; Ma, D.-L. Inhibition of Beta-Amyloid Fibrillation by Luminescent Iridium(III) Complex Probes. Sci. Rep. 2015, 5, 14619. [Google Scholar] [CrossRef]
- Barnham, K.J.; Kenche, V.B.; Ciccotosto, G.D.; Smith, D.P.; Tew, D.J.; Liu, X.; Perez, K.; Cranston, G.A.; Johanssen, T.J.; Volitakis, I.; et al. Platinum-based inhibitors of amyloid-β as therapeutic agents for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 6813–6818. [Google Scholar] [CrossRef]
- Kenche, V.B.; Hung, L.W.; Perez, K.; Volitakes, I.; Ciccotosto, G.; Kwok, J.; Critch, N.; Sherratt, N.; Cortes, M.; Lal, V.; et al. Development of a Platinum Complex as an anti-Amyloid Agent for the Therapy of Alzheimer’s Disease. Angew. Chem. Int. Ed. 2013, 52, 3374–3378. [Google Scholar] [CrossRef]
- Franz, K.J. Application of inorganic chemistry for non-cancer therapeutics. Dalt. Trans. 2012, 41, 6333–6334. [Google Scholar] [CrossRef] [PubMed]
- Streltsov, V.A.; Chandana Epa, V.; James, S.A.; Churches, Q.I.; Caine, J.M.; Kenche, V.B.; Barnham, K.J. Structural insights into the interaction of platinum-based inhibitors with the Alzheimer’s disease amyloid-β peptide. Chem. Commun. 2013, 49, 11364–11366. [Google Scholar] [CrossRef]
- Avan, A.; Postma, T.J.; Ceresa, C.; Avan, A.; Cavaletti, G.; Giovannetti, E.; Peters, G.J. Platinum-Induced Neurotoxicity and Preventive Strategies: Past, Present, and Future. Oncologist 2015, 20, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Howson, S.E.; Dong, K.; Gao, N.; Ren, J.; Scott, P.; Qu, X. Chiral metallohelical complexes enantioselectively target amyloid β for Treating Alzheimer’s disease. J. Am. Chem. Soc. 2014, 136, 11655–11663. [Google Scholar] [CrossRef]
- Chen, W.; Ouyang, J.; Yi, X.; Xu, Y.; Niu, C.; Zhang, W.; Wang, L.; Sheng, J.; Deng, L.; Liu, Y.-N.; et al. Black Phosphorus Nanosheets as a Neuroprotective Nanomedicine for Neurodegenerative Disorder Therapy. Adv. Mater. 2018, 30, 1703458. [Google Scholar] [CrossRef]
- Mir J, M. Design of Metal Complexes as Anti-AD Agents. In Alzheimer’s Disease & Treatment; MedDocs Publishers LLC: Reno, NV, USA, 2017; p. 7. ISBN 978-81-936678-1-1. [Google Scholar]
- Jalili-Baleh, L.; Nadri, H.; Forootanfar, H.; Samzadeh-Kermani, A.; Küçükkılınç, T.T.; Ayazgok, B.; Rahimifard, M.; Baeeri, M.; Doostmohammadi, M.; Firoozpour, L.; et al. Novel 3-phenylcoumarin–lipoic acid conjugates as multi-functional agents for potential treatment of Alzheimer’s disease. Bioorg. Chem. 2018, 79, 223–234. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, H.; Li, X.; Dong, S.; Liu, W.; Gong, Q.; Wang, T.; Tang, Y.; Zhu, J.; Li, J.; et al. Discovery of novel propargylamine-modified 4-aminoalkyl imidazole substituted pyrimidinylthiourea derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 143, 33–47. [Google Scholar] [CrossRef]
- Grasso, G.; Santoro, A.M.; Lanza, V.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Marini, S.; Coletta, M.; Milardi, D. The double faced role of copper in Aβ homeostasis: A survey on the interrelationship between metal dyshomeostasis, UPS functioning and autophagy in neurodegeneration. Coord. Chem. Rev. 2017, 347, 1–22. [Google Scholar] [CrossRef]
- Rana, M.; Cho, H.-J.; Roy, T.K.; Mirica, L.M.; Sharma, A.K. Azo-dyes based small bifunctional molecules for metal chelation and controlling amyloid formation. Inorganica Chim. Acta 2018, 471, 419–429. [Google Scholar] [CrossRef]
- Wang, X.-Q.; Zhao, C.-P.; Zhong, L.-C.; Zhu, D.-L.; Mai, D.-H.; Liang, M.-G.; He, M.-H. Preparation of 4-Flexible Amino-2-Arylethenyl-Quinoline Derivatives as Multi-target Agents for the Treatment of Alzheimer’s Disease. Molecules 2018, 23, 3100. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S. 8-Hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Devel. Ther. 2013, 7, 1157. [Google Scholar] [CrossRef]
- Ono, M.; Watanabe, H.; Watanabe, R.; Haratake, M.; Nakayama, M.; Saji, H. Diphenylpropynone derivatives as probes for imaging β-amyloid plaques in Alzheimer’s brains. Bioorg. Med. Chem. Lett. 2011, 21, 117–120. [Google Scholar] [CrossRef]
- Jones, M.R.; Mathieu, E.; Dyrager, C.; Faissner, S.; Vaillancourt, Z.; Korshavn, K.J.; Lim, M.H.; Ramamoorthy, A.; Wee Yong, V.; Tsutsui, S.; et al. Multi-target-directed phenol–triazole ligands as therapeutic agents for Alzheimer’s disease. Chem. Sci. 2017, 8, 5636–5643. [Google Scholar] [CrossRef]
- Zhang, C.; Gomes, L.M.F.; Zhang, T.; Storr, T. A small bifunctional chelator that modulates Aβ42 aggregation. Can. J. Chem. 2018, 96, 78–82. [Google Scholar] [CrossRef]
- Liu, Y.; Kochi, A.; Pithadia, A.S.; Lee, S.; Nam, Y.; Beck, M.W.; He, X.; Lee, D.; Lim, M.H. Tuning Reactivity of Diphenylpropynone Derivatives with Metal-Associated Amyloid-β Species via Structural Modifications. Inorg. Chem. 2013, 52, 8121–8130. [Google Scholar] [CrossRef]
- Cao, Z.; Yang, J.; Xu, R.; Song, Q.; Zhang, X.; Liu, H.; Qiang, X.; Li, Y.; Tan, Z.; Deng, Y. Design, synthesis and evaluation of 4′-OH-flurbiprofen-chalcone hybrids as potential multifunctional agents for Alzheimer’s disease treatment. Bioorg. Med. Chem. 2018, 26, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Fosso, M.Y.; LeVine, H., 3rd; Green, K.D.; Tsodikov, O.V.; Garneau-Tsodikova, S. Effects of structural modifications on the metal binding, anti-amyloid activity, and cholinesterase inhibitory activity of chalcones. Org. Biomol. Chem. 2015, 13, 9418–9426. [Google Scholar] [CrossRef]
- Schugar, H.; Green, D.E.; Bowen, M.L.; Scott, L.E.; Storr, T.; Böhmerle, K.; Thomas, F.; Allen, D.D.; Lockman, P.R.; Merkel, M.; et al. Combating Alzheimer’s Disease With Multifunctional Molecules Designed for Metal Passivation. Angew. Chem. Int. Ed. 2007, 46, 1716–1718. [Google Scholar] [CrossRef] [PubMed]
- Telpoukhovskaia, M.A.; Cawthray, J.F.; Rodríguez-Rodríguez, C.; Scott, L.E.; Page, B.D.G.; Patrick, B.O.; Orvig, C. 3-Hydroxy-4-pyridinone derivatives designed for fluorescence studies to determine interaction with amyloid protein as well as cell permeability. Bioorg. Med. Chem. Lett. 2015, 25, 3654–3657. [Google Scholar] [CrossRef]
- Telpoukhovskaia, M.A.; Rodríguez-Rodríguez, C.; Cawthray, J.F.; Scott, L.E.; Page, B.D.G.; Alí-Torres, J.; Sodupe, M.; Bailey, G.A.; Patrick, B.O.; Orvig, C. 3-Hydroxy-4-pyridinone derivatives as metal ion and amyloid binding agents. Metallomics 2014, 6, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Bowen, M.L.; Scott, L.E.; Storr, T.; Merkel, M.; Böhmerle, K.; Thompson, K.H.; Patrick, B.O.; Schugar, H.J.; Orvig, C. In vitro studies of 3-hydroxy-4-pyridinones and their glycosylated derivatives as potential agents for Alzheimer’s disease. Dalt. Trans. 2010, 39, 1604–1615. [Google Scholar] [CrossRef]
- Yang, X.; Cai, P.; Liu, Q.; Wu, J.; Yin, Y.; Wang, X.; Kong, L. Novel 8-hydroxyquinoline derivatives targeting β -amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorg. Med. Chem. 2018, 26, 3191–3201. [Google Scholar] [CrossRef]
- Gomes, L.M.F.; Vieira, R.P.; Jones, M.R.; Wang, M.C.P.; Dyrager, C.; Souza-Fagundes, E.M.; Da Silva, J.G.; Storr, T.; Beraldo, H. 8-Hydroxyquinoline Schiff-base compounds as antioxidants and modulators of copper-mediated Aβ peptide aggregation. J. Inorg. Biochem. 2014, 139, 106–116. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, J.; Yang, X.; Feng, X.; Li, X.; Huang, L.; Chan, A.S.C. Design, Synthesis, and Evaluation of Orally Bioavailable Quinoline–Indole Derivatives as Innovative Multitarget-Directed Ligands: Promotion of Cell Proliferation in the Adult Murine Hippocampus for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2018, 61, 1871–1894. [Google Scholar] [CrossRef]
- Zheng, H.; Youdim, M.B.H.; Fridkin, M. Site-Activated Multifunctional Chelator with Acetylcholinesterase and Neuroprotective−Neurorestorative Moieties for Alzheimer’s Therapy. J. Med. Chem. 2009, 52, 4095–4098. [Google Scholar] [CrossRef]
- Oliveri, V.; Grasso, G.I.; Bellia, F.; Attanasio, F.; Viale, M.; Vecchio, G. Soluble Sugar-Based Quinoline Derivatives as New Antioxidant Modulators of Metal-Induced Amyloid Aggregation. Inorg. Chem. 2015, 54, 2591–2602. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, T.; Zhu, S.; Gu, X.; Jia, X.; Lu, Y.; Zhu, L. Two macrocyclic polyamines as modulators of metal-mediated Aβ40 aggregation. Integr. Biol. 2015, 7, 655–662. [Google Scholar] [CrossRef]
- Lanza, V.; D’Agata, R.; Iacono, G.; Bellia, F.; Spoto, G.; Vecchio, G. Cyclam glycoconjugates as lectin ligands and protective agents of metal-induced amyloid aggregation. J. Inorg. Biochem. 2015, 153, 377–382. [Google Scholar] [CrossRef]
- Lincoln, K.M.; Gonzalez, P.; Richardson, T.E.; Julovich, D.A.; Saunders, R.; Simpkins, J.W.; Green, K.N. A potent antioxidant small molecule aimed at targeting metal-based oxidative stress in neurodegenerative disorders. Chem. Commun. 2013, 49, 2712. [Google Scholar] [CrossRef]
- Lincoln, K.M.; Richardson, T.E.; Rutter, L.; Gonzalez, P.; Simpkins, J.W.; Green, K.N. An N-Heterocyclic Amine Chelate Capable of Antioxidant Capacity and Amyloid Disaggregation. ACS Chem. Neurosci. 2012, 3, 919–927. [Google Scholar] [CrossRef]
- Gonzalez, P.; da Costa, V.C.P.; Hyde, K.; Wu, Q.; Annunziata, O.; Rizo, J.; Akkaraju, G.; Green, K.N. Bimodal-hybrid heterocyclic amine targeting oxidative pathways and copper mis-regulation in Alzheimer’s disease. Metallomics 2014, 6, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Mu, C.; Wang, M.C.P.; Webb, M.I.; Walsby, C.J.; Storr, T. Modulation of the Aβ peptide aggregation pathway by KP1019 limits Aβ-associated neurotoxicity. Metallomics 2015, 7, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.-Y.; Yin, W.-X.; Yin, J.; Zhang, S.-B.; Liu, C.-L. The chelation targeting metal–Aβ40 aggregates may lead to formation of Aβ40 oligomers. Dalt. Trans. 2011, 40, 4830. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, C.; Telpoukhovskaia, M.A.; Alí-Torres, J.; Rodríguez-Santiago, L.; Manso, Y.; Bailey, G.A.; Hidalgo, J.; Sodupe, M.; Orvig, C. Thioflavin-based molecular probes for application in Alzheimer’s disease: from in silico to in vitro models. Metallomics 2015, 7, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, C.; SáNchez De Groot, N.; Rimola, A.; Álvarez-Larena, Á.; Lloveras, V.; Vidal-Gancedo, J.; Ventura, S.; Vendrell, J.; Sodupe, M.; GonzáLez-Duarte, P. Design, Selection, and Characterization of Thioflavin-Based Intercalation Compounds with Metal Chelating Properties for Application in Alzheimer’s Disease. J. Am. Chem. Soc. 2009, 131, 1436–1451. [Google Scholar]
- Hindo, S.S.; Mancino, A.M.; Braymer, J.J.; Liu, Y.; Vivekanandan, S.; Ramamoorthy, A.; Lim, M.H. Small Molecule Modulators of Copper-Induced Aβ Aggregation. J. Am. Chem. Soc. 2009, 131, 16663–16665. [Google Scholar] [CrossRef]
- Viveiros, R.; Karim, K.; Piletsky, S.A.; Heggie, W.; Casimiro, T. Development of a molecularly imprinted polymer for a pharmaceutical impurity in supercritical CO2: Rational design using computational approach. J. Clean. Prod. 2017, 168, 1025–1031. [Google Scholar] [CrossRef]
- Braymer, J.J.; Choi, J.-S.; DeToma, A.S.; Wang, C.; Nam, K.; Kampf, J.W.; Ramamoorthy, A.; Lim, M.H. Development of Bifunctional Stilbene Derivatives for Targeting and Modulating Metal-Amyloid-β Species. Inorg. Chem. 2011, 50, 10724–10734. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Ko, K.S.; Stinnett, H.; Van der Schyf, C.J.; Lim, M.H. Identification of multifunctional small molecule-based reversible monoamine oxidase inhibitors. Medchemcomm 2011, 2, 1099. [Google Scholar] [CrossRef]
- Choi, J.-S.; Braymer, J.J.; Nanga, R.P.R.; Ramamoorthy, A.; Lim, M.H. Design of small molecules that target metal-A{beta} species and regulate metal-induced A{beta} aggregation and neurotoxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 21990–21995. [Google Scholar] [CrossRef]
- Beck, M.W.; Derrick, J.S.; Kerr, R.A.; Oh, S.B.; Cho, W.J.; Lee, S.J.C.; Ji, Y.; Han, J.; Tehrani, Z.A.; Suh, N.; et al. Structure-mechanism-based engineering of chemical regulators targeting distinct pathological factors in Alzheimer’s disease. Nat. Commun. 2016, 7, 13115. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, X.-B.; Li, Z.-R.; Li, S.-Y.; Xie, S.-S.; Huang, M.; Kong, L.-Y. Design of a structural framework with potential use to develop balanced multifunctional agents against Alzheimer’s disease. RSC Adv. 2015, 5, 14242–14255. [Google Scholar] [CrossRef]
- Lee, S.; Zheng, X.; Krishnamoorthy, J.; Savelieff, M.G.; Park, H.M.; Brender, J.R.; Kim, J.H.; Derrick, J.S.; Kochi, A.; Lee, H.J.; et al. Rational Design of a Structural Framework with Potential Use to Develop Chemical Reagents That Target and Modulate Multiple Facets of Alzheimer’s Disease. J. Am. Chem. Soc. 2014, 136, 299–310. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Liu, Y.; Senthamarai, R.R.P.; Korshavn, K.J.; Lee, H.J.; Ramamoorthy, A.; Lim, M.H. A small molecule that displays marked reactivity toward copper- versus zinc-amyloid-β implicated in Alzheimer’s disease. Chem. Commun. 2014, 50, 5301–5303. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, M.; Sheng, R.; Ma, Y. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ 1–42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 174–186. [Google Scholar] [CrossRef]
- Li, S.-Y.; Wang, X.-B.; Kong, L.-Y. Design, synthesis and biological evaluation of imine resveratrol derivatives as multi-targeted agents against Alzheimer’s disease. Eur. J. Med. Chem. 2014, 71, 36–45. [Google Scholar] [CrossRef]
- DeToma, A.S.; Krishnamoorthy, J.; Nam, Y.; Lee, H.J.; Brender, J.R.; Kochi, A.; Lee, D.; Onnis, V.; Congiu, C.; Manfredini, S.; et al. Interaction and reactivity of synthetic aminoisoflavones with metal-free and metal-associated amyloid-β. Chem. Sci. 2014, 5, 4851–4862. [Google Scholar] [CrossRef]
- He, X.; Park, H.M.; Hyung, S.-J.; DeToma, A.S.; Kim, C.; Ruotolo, B.T.; Lim, M.H. Exploring the reactivity of flavonoid compounds with metal-associated amyloid-β species. Dalt. Trans. 2012, 41, 6558. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Youdim, M.B.H.; Fridkin, M. Selective Acetylcholinesterase Inhibitor Activated by Acetylcholinesterase Releases an Active Chelator with Neurorescuing and Anti-Amyloid Activities. ACS Chem. Neurosci. 2010, 1, 737–746. [Google Scholar] [CrossRef]
- Wang, Z.-M.; Cai, P.; Liu, Q.-H.; Xu, D.-Q.; Yang, X.-L.; Wu, J.-J.; Kong, L.-Y.; Wang, X.-B. Rational modification of donepezil as multifunctional acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2016, 123, 282–297. [Google Scholar] [CrossRef]
- Li, F.; Wang, Z.-M.; Wu, J.-J.; Wang, J.; Xie, S.-S.; Lan, J.-S.; Xu, W.; Kong, L.-Y.; Wang, X.-B. Synthesis and pharmacological evaluation of donepezil-based agents as new cholinesterase/monoamine oxidase inhibitors for the potential application against Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2016, 31, 41–53. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Esteban, G.; Brogi, S.; Shionoya, M.; Wang, L.; Campiani, G.; Unzeta, M.; Inokuchi, T.; Butini, S.; Marco-Contelles, J. Donepezil-like multifunctional agents: Design, synthesis, molecular modeling and biological evaluation. Eur. J. Med. Chem. 2016, 121, 864–879. [Google Scholar] [CrossRef]
- Wang, L.; Esteban, G.; Ojima, M.; Bautista-Aguilera, O.M.; Inokuchi, T.; Moraleda, I.; Iriepa, I.; Samadi, A.; Youdim, M.B.H.; Romero, A.; et al. Donepezil + propargylamine + 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 80, 543–561. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Valgimigli, L.; Bartolini, M.; Rosini, M.; Andrisano, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Drug Design Strategy: From a Dual Binding Site Acetylcholinesterase Inhibitor to a Trifunctional Compound against Alzheimer’s Disease. J. Med. Chem. 2007, 50, 6446–6449. [Google Scholar] [CrossRef]
- Hui, A.; Chen, Y.; Zhu, S.; Gan, C.; Pan, J.; Zhou, A. Design and synthesis of tacrine-phenothiazine hybrids as multitarget drugs for Alzheimer’s disease. Med. Chem. Res. 2014, 23, 3546–3557. [Google Scholar] [CrossRef]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget Drug Design Strategy: Quinone–Tacrine Hybrids Designed To Block Amyloid-β Aggregation and To Exert Anticholinesterase and Antioxidant Effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef]
- Fang, L.; Kraus, B.; Lehmann, J.; Heilmann, J.; Zhang, Y.; Decker, M. Design and synthesis of tacrine–ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 2905–2909. [Google Scholar] [CrossRef]
- Benchekroun, M.; Romero, A.; Egea, J.; León, R.; Michalska, P.; Buendía, I.; Jimeno, M.L.; Jun, D.; Janockova, J.; Sepsova, V.; et al. The Antioxidant Additive Approach for Alzheimer’s Disease Therapy: New Ferulic (Lipoic) Acid Plus Melatonin Modified Tacrines as Cholinesterases Inhibitors, Direct Antioxidants, and Nuclear Factor (Erythroid-Derived 2)-Like 2 Activators. J. Med. Chem. 2016, 59, 9967–9973. [Google Scholar] [CrossRef] [PubMed]
- Benchekroun, M.; Bartolini, M.; Egea, J.; Romero, A.; Soriano, E.; Pudlo, M.; Luzet, V.; Andrisano, V.; Jimeno, M.-L.; López, M.G.; et al. Novel Tacrine-Grafted Ugi Adducts as Multipotent Anti-Alzheimer Drugs: A Synthetic Renewal in Tacrine-Ferulic Acid Hybrids. ChemMedChem 2015, 10, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel Tacrine−8-Hydroxyquinoline Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease, with Neuroprotective, Cholinergic, Antioxidant, and Copper-Complexing Properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Hepnarova, V.; Korabecny, J.; Matouskova, L.; Jost, P.; Muckova, L.; Hrabinova, M.; Vykoukalova, N.; Kerhartova, M.; Kucera, T.; Dolezal, R.; et al. The concept of hybrid molecules of tacrine and benzyl quinolone carboxylic acid (BQCA) as multifunctional agents for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 292–306. [Google Scholar] [CrossRef]
- Antequera, D.; Bolos, M.; Spuch, C.; Pascual, C.; Ferrer, I.; Fernandez-Bachiller, M.I.; Rodríguez-Franco, M.I.; Carro, E. Effects of a tacrine-8-hydroxyquinoline hybrid (IQM-622) on Aβ accumulation and cell death: Involvement in hippocampal neuronal loss in Alzheimer’s disease. Neurobiol. Dis. 2012, 46, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Skibiński, R.; Czarnecka, K.; Girek, M.; Bilichowski, I.; Chufarova, N.; Mikiciuk-Olasik, E.; Szymański, P. Novel tetrahydroacridine derivatives with iodobenzoic acid moiety as multifunctional acetylcholinesterase inhibitors. Chem. Biol. Drug Des. 2018, 91, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Huang, L.; Luo, Z.; Liu, A.; Lu, C.; Xie, Z.; Li, X. O-Hydroxyl- or o-amino benzylamine-tacrine hybrids: Multifunctional biometals chelators, antioxidants, and inhibitors of cholinesterase activity and amyloid-β aggregation. Bioorg. Med. Chem. 2012, 20, 5884–5892. [Google Scholar] [CrossRef]
- Dgachi, Y.; Sokolov, O.; Luzet, V.; Godyń, J.; Panek, D.; Bonet, A.; Martin, H.; Iriepa, I.; Moraleda, I.; García-Iriepa, C.; et al. Tetrahydropyranodiquinolin-8-amines as new, non hepatotoxic, antioxidant, and acetylcholinesterase inhibitors for Alzheimer’s disease therapy. Eur. J. Med. Chem. 2017, 126, 576–589. [Google Scholar] [CrossRef]
- Li, S.-Y.; Wang, X.-B.; Xie, S.-S.; Jiang, N.; Wang, K.D.G.; Yao, H.-Q.; Sun, H.-B.; Kong, L.-Y. Multifunctional tacrine–flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2013, 69, 632–646. [Google Scholar] [CrossRef]
- Sun, Q.; Peng, D.-Y.; Yang, S.-G.; Zhu, X.-L.; Yang, W.-C.; Yang, G.-F. Syntheses of coumarin–tacrine hybrids as dual-site acetylcholinesterase inhibitors and their activity against butylcholinesterase, Aβ aggregation, and β-secretase. Bioorg. Med. Chem. 2014, 22, 4784–4791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jin, B.; Shi, Z.; Wang, X.; Lei, S.; Tang, X.; Liang, H.; Liu, Q.; Gong, M.; Peng, R. New tris(dopamine) derivative as an iron chelator. Synthesis, solution thermodynamic stability, and antioxidant research. J. Inorg. Biochem. 2017, 171, 29–36. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Madhu, C.; Govindaraju, T. Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid β Toxicity. ACS Chem. Neurosci. 2016, 7, 1300–1310. [Google Scholar] [CrossRef]
- Trapaidze, A.; Hureau, C.; Bal, W.; Winterhalter, M.; Faller, P. Thermodynamic study of Cu2+ binding to the DAHK and GHK peptides by isothermal titration calorimetry (ITC) with the weaker competitor glycine. JBIC J. Biol. Inorg. Chem. 2012, 17, 37–47. [Google Scholar] [CrossRef]
- Márquez, M.; Blancas-Mejía, L.M.; Campos, A.; Rojas, L.; Castañeda-Hernández, G.; Quintanar, L. A bifunctional non-natural tetrapeptide modulates amyloid-beta peptide aggregation in the presence of Cu(ii). Metallomics 2014, 6, 2189–2192. [Google Scholar] [CrossRef] [PubMed]
- McCabe, J.W.; Vangala, R.; Angel, L.A. Binding Selectivity of Methanobactin from Methylosinus trichosporium OB3b for Copper(I), Silver(I), Zinc(II), Nickel(II), Cobalt(II), Manganese(II), Lead(II), and Iron(II). J. Am. Soc. Mass Spectrom. 2017, 28, 2588–2601. [Google Scholar] [CrossRef]
- Iraji, A.; Firuzi, O.; Khoshneviszadeh, M.; Tavakkoli, M.; Mahdavi, M.; Nadri, H.; Edraki, N.; Miri, R. Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur. J. Med. Chem. 2017, 141, 690–702. [Google Scholar] [CrossRef]
- De Simone, A.; Bartolini, M.; Baschieri, A.; Apperley, K.Y.P.; Chen, H.H.; Guardigni, M.; Montanari, S.; Kobrlova, T.; Soukup, O.; Valgimigli, L.; et al. Hydroxy-substituted trans-cinnamoyl derivatives as multifunctional tools in the context of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 139, 378–389. [Google Scholar] [CrossRef]
- Hayne, D.J.; Lim, S.; Donnelly, P.S. Metal complexes designed to bind to amyloid-β for the diagnosis and treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6701–6715. [Google Scholar] [CrossRef]
- Kim, I.; Kim, C.H.; Kim, J.H.; Lee, J.; Choi, J.J.; Chen, Z.A.; Lee, M.G.; Chung, K.C.; Hsu, C.Y.; Ahn, Y.S. Pyrrolidine dithiocarbamate and zinc inhibit proteasome-dependent proteolysis. Exp. Cell Res. 2004, 298, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Thom, V.J.; Hosken, G.D.; Hancock, R. Anomalous Metal Ion Size Selectivity of Tetraaza Macrocycles. Inorg. Chem 1985, 24, 33783381. [Google Scholar]
- Sharma, A.; Pachauri, V.; Flora, S.J.S. Advances in Multi-Functional Ligands and the Need for Metal-Related Pharmacology for the Management of Alzheimer Disease. Front. Pharmacol. 2018, 9, 1247. [Google Scholar] [CrossRef] [PubMed]
- Lanza, V.; Milardi, D.; Di Natale, G.; Pappalardo, G. Repurposing of Copper(II)-chelating Drugs for the Treatment of Neurodegenerative Diseases. Curr. Med. Chem. 2018, 25, 525–539. [Google Scholar] [CrossRef]
- Kim, T.-W. Drug Repositioning Approaches for the Discovery of New Therapeutics for Alzheimer’s Disease. Neurotherapeutics 2015, 12, 132–142. [Google Scholar] [CrossRef]
- Durães, F.; Pinto, M.; Sousa, E.; Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef]
- Mucke, H.A. The case of galantamine: repurposing and late blooming of a cholinergic drug. Futur. Sci. OA 2015, 1, FSO73. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.D.; Dey, D.; Palavicini, J.P.; Wang, H.; Patkar, K.A.; Minond, D.; Nefzi, A.; Lakshmana, M.K. Striking reduction of amyloid plaque burden in an Alzheimer’s mouse model after chronic administration of carmustine. BMC Med. 2013, 11, 81. [Google Scholar] [CrossRef]
- Fukasawa, H.; Nakagomi, M.; Yamagata, N.; Katsuki, H.; Kawahara, K.; Kitaoka, K.; Miki, T.; Shudo, K. Tamibarotene: A Candidate Retinoid Drug for Alzheimer’s Disease. Biol. Pharm. Bull. 2012, 35, 1206–1212. [Google Scholar] [CrossRef]
- Netzer, W.J.; Dou, F.; Cai, D.; Veach, D.; Jean, S.; Li, Y.; Bornmann, W.G.; Clarkson, B.; Xu, H.; Greengard, P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. USA 2003, 100, 12444–12449. [Google Scholar] [CrossRef] [PubMed]
- Tousi, B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: current evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311. [Google Scholar] [PubMed]
- Brunden, K.R.; Yao, Y.; Potuzak, J.S.; Ferrer, N.I.; Ballatore, C.; James, M.J.; Hogan, A.-M.L.; Trojanowski, J.Q.; Smith, A.B.; Lee, V.M.-Y. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol. Res. 2011, 63, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-α in an animal model of inflamed Alzheimer’s disease brain. Neurobiol. Dis. 2008, 29, 254–266. [Google Scholar] [CrossRef]
- Diomede, L.; Cassata, G.; Fiordaliso, F.; Salio, M.; Ami, D.; Natalello, A.; Doglia, S.M.; De Luigi, A.; Salmona, M. Tetracycline and its analogues protect Caenorhabditis elegans from β amyloid-induced toxicity by targeting oligomers. Neurobiol. Dis. 2010, 40, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Gaeta, A.; Francis, P.T.; Williams, R.J.; Hider, R.C. Neuroprotective actions of deferiprone in cultured cortical neurones and SHSY-5Y cells. J. Neurochem. 2008, 105, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, G.; Cossu, G.; Balocco, M.; Marchese, R.; Murgia, D.; Melis, M.; Galanello, R.; Barella, S.; Matta, G.; Ruffinengo, U.; et al. A pilot trial of deferiprone for neurodegeneration with brain iron accumulation. Haematologica 2011, 96, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Fredenburg, A.M.; Sethi, R.K.; Allen, D.D.; Yokel, R.A. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1’ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology 1996, 108, 191–199. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Drug Repurposing in Parkinson’s Disease. CNS Drugs 2018, 32, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.-S.; Mitterstiller, A.-M.; Breuer, W.; Weiss, G.; Cabantchik, Z.I. Rescuing iron-overloaded macrophages by conservative relocation of the accumulated metal. Br. J. Pharmacol. 2011, 164, 406–418. [Google Scholar] [CrossRef]
- Li, S.-J.; Qin, W.-X.; Peng, D.-J.; Yuan, Z.-X.; He, S.-N.; Luo, Y.-N.; Aschner, M.; Jiang, Y.-M.; Liang, D.-Y.; Xie, B.-Y.; et al. Sodium P -aminosalicylic acid inhibits sub-chronic manganese-induced neuroinflammation in rats by modulating MAPK and COX-2. Neurotoxicology 2018, 64, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, M.; Eap, C.B. Pharmacodynamic, Pharmacokinetic and Pharmacogenetic Aspects of Drugs Used in the Treatment of Alzheimer’s Disease. Clin. Pharmacokinet. 2013, 52, 225–241. [Google Scholar] [CrossRef]
- Noetzli, M.; Guidi, M.; Ebbing, K.; Eyer, S.; Zumbach, S.; Giannakopoulos, P.; von Gunten, A.; Csajka, C.; Eap, C.B. Relationship of CYP2D6, CYP3A, POR, and ABCB1 Genotypes With Galantamine Plasma Concentrations. Ther. Drug Monit. 2013, 35, 270–275. [Google Scholar] [CrossRef]
- Chianella, C.; Gragnaniello, D.; Maisano Delser, P.; Visentini, M.F.; Sette, E.; Tola, M.R.; Barbujani, G.; Fuselli, S. BCHE and CYP2D6 genetic variation in Alzheimer’s disease patients treated with cholinesterase inhibitors. Eur. J. Clin. Pharmacol. 2011, 67, 1147–1157. [Google Scholar] [CrossRef]
- Pilotto, A.; Franceschi, M.; D’Onofrio, G.; Bizzarro, A.; Mangialasche, F.; Cascavilla, L.; Paris, F.; Matera, M.G.; Pilotto, A.; Daniele, A.; et al. Effect of a CYP2D6 polymorphism on the efficacy of donepezil in patients with Alzheimer disease. Neurology 2009, 73, 761–767. [Google Scholar] [CrossRef]
- Varsaldi, F.; Miglio, G.; Scordo, M.G.; Dahl, M.-L.; Villa, L.M.; Biolcati, A.; Lombardi, G. Impact of the CYP2D6 polymorphism on steady-state plasma concentrations and clinical outcome of donepezil in Alzheimer’s disease patients. Eur. J. Clin. Pharmacol. 2006, 62, 721–726. [Google Scholar] [CrossRef]
- Seripa, D.; Bizzarro, A.; Pilotto, A.; DʼOnofrio, G.; Vecchione, G.; Gallo, A.P.; Cascavilla, L.; Paris, F.; Grandone, E.; Mecocci, P.; et al. Role of cytochrome P4502D6 functional polymorphisms in the efficacy of donepezil in patients with Alzheimerʼs disease. Pharmacogenet. Genom. 2010, 21, 1. [Google Scholar] [CrossRef]
- Makhtar, S.M.; Husin, A.; Baba, A.A.; Ankathil, R. Genetic variations in influx transporter gene SLC22A1 are associated with clinical responses to imatinib mesylate among Malaysian chronic myeloid leukaemia patients. J. Genet. 2018, 97, 835–842. [Google Scholar] [CrossRef]
- Ben Hassine, I.; Gharbi, H.; Soltani, I.; Ben Hadj Othman, H.; Farrah, A.; Amouri, H.; Teber, M.; Ghedira, H.; Ben Youssef, Y.; Safra, I.; et al. Molecular study of ABCB1 gene and its correlation with imatinib response in chronic myeloid leukemia. Cancer Chemother. Pharmacol. 2017, 80, 829–839. [Google Scholar] [CrossRef]
- Andriguetti, N.B.; Raymundo, S.; Antunes, M.V.; Perassolo, M.S.; Verza, S.G.; Suyenaga, E.S.; Linden, R. Pharmacogenetic and Pharmacokinetic Dose Individualization of the Taxane Chemotherapeutic Drugs Paclitaxel and Docetaxel. Curr. Med. Chem. 2017, 24. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Pauli-Magnus, C.; Hodges, L.M.; Huang, C.C.; Kawamoto, M.; Johns, S.J.; Stryke, D.; Ferrin, T.E.; DeYoung, J.; Taylor, T.; et al. Sequence diversity and haplotype structure in the human ABCB1 (MDR1, multidrug resistance transporter) gene. Pharmacogenetics 2003, 13, 481–494. [Google Scholar] [CrossRef]
- Dadheech, S.; Rao, A.V.; Shaheen, U.; Hussien, M.D.; Jain, S.; Jyothy, A.; Munshi, A. Three most common nonsynonymous UGT1A6*2 polymorphisms (Thr181Ala, Arg184Serand Ser7Ala) and therapeutic response to deferiprone in β-thalassemia major patients. Gene 2013, 531, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.W.; Bush, A.I.; Masters, C.L. Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin. Investig. Drugs 2004, 13, 1585–1592. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Matlack, K.E.S.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid- (A) oligomers to restore endocytosis and ameliorate A toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A. Prophylactic value of clioquinol against travellers’ diarrhoea. Lancet 1971, 1, 44–45. [Google Scholar]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Zatta, P.; Drago, D.; Bolognin, S.; Sensi, S.L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, C.; Telpoukhovskaia, M.; Orvig, C. The art of building multifunctional metal-binding agents from basic molecular scaffolds for the potential application in neurodegenerative diseases. Coord. Chem. Rev. 2012, 256, 2308–2332. [Google Scholar]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.-X.; Tammer, A.; et al. Metal-Protein Attenuation With Iodochlorhydroxyquin (Clioquinol) Targeting Aβ Amyloid Deposition and Toxicity in Alzheimer Disease. Arch. Neurol. 2003, 60, 1685. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Raymick, J.; Sarkar, S.; Lahiri, D.K.; Ray, B.; Holtzman, D.; Dumas, M.; Schmued, L.C. Efficacy and toxicity of clioquinol treatment and A-beta42 inoculation in the APP/PSI mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 494–506. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 Rapidly Improves Cognition in Alzheimer’s Disease: Additional Phase II Analyses. J. Alzheimer’s Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar]
- Cukierman, D.S.; Pinheiro, A.B.; Castiñeiras-Filho, S.L.P.; da Silva, A.S.P.; Miotto, M.C.; De Falco, A.; de P Ribeiro, T.; Maisonette, S.; da Cunha, A.L.M.C.; Hauser-Davis, R.A.; et al. A moderate metal-binding hydrazone meets the criteria for a bioinorganic approach towards Parkinson’s disease: Therapeutic potential, blood-brain barrier crossing evaluation and preliminary toxicological studies. J. Inorg. Biochem. 2017, 170, 160–168. [Google Scholar]
- Hauser-Davis, R.A.; de Freitas, L.V.; Cukierman, D.S.; Cruz, W.S.; Miotto, M.C.; Landeira-Fernandez, J.; Valiente-Gabioud, A.A.; Fernández, C.O.; Rey, N.A. Disruption of zinc and copper interactions with Aβ(1–40) by a non-toxic, isoniazid-derived, hydrazone: A novel biometal homeostasis restoring agent in Alzheimer’s disease therapy? Metallomics 2015, 7, 743–747. [Google Scholar] [CrossRef]
- Cukierman, D.S.; Accardo, E.; Gomes, R.G.; De Falco, A.; Miotto, M.C.; Freitas, M.C.R.; Lanznaster, M.; Fernández, C.O.; Rey, N.A. Aroylhydrazones constitute a promising class of ‘metal-protein attenuating compounds’ for the treatment of Alzheimer’s disease: A proof-of-concept based on the study of the interactions between zinc(II) and pyridine-2-carboxaldehyde isonicotinoyl hydrazone. JBIC J. Biol. Inorg. Chem. 2018, 23, 1227–1241. [Google Scholar] [CrossRef]
- Ji, H.-F.; Zhang, H.-Y. A new strategy to combat Alzheimer’s disease. Combining radical-scavenging potential with metal-protein-attenuating ability in one molecule. Bioorg. Med. Chem. Lett. 2005, 15, 21–24. [Google Scholar] [CrossRef]
- Kuca, K.; Soukup, O.; Maresova, P.; Korabecny, J.; Nepovimova, E.; Klimova, B.; Honegr, J.; Ramalho, T.C.; França, T.C.C. Current Approaches Against Alzheimer’s Disease in Clinical Trials. J. Braz. Chem. Soc. 2016, 27, 641–649. [Google Scholar] [CrossRef]
- Gonçalves, A.S.; França, T.C.C.; Caetano, M.S.; Ramalho, T.C. Reactivation steps by 2-PAM of tabun-inhibited human acetylcholinesterase: Reducing the computational cost in hybrid QM/MM methods. J. Biom. Struct. Dyn. 2014, 32, 301–307. [Google Scholar]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sales, T.A.; Prandi, I.G.; de Castro, A.A.; Leal, D.H.S.; da Cunha, E.F.F.; Kuca, K.; Ramalho, T.C. Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. Int. J. Mol. Sci. 2019, 20, 1829. https://doi.org/10.3390/ijms20081829
Sales TA, Prandi IG, de Castro AA, Leal DHS, da Cunha EFF, Kuca K, Ramalho TC. Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. International Journal of Molecular Sciences. 2019; 20(8):1829. https://doi.org/10.3390/ijms20081829
Chicago/Turabian StyleSales, Thais A., Ingrid G. Prandi, Alexandre A. de Castro, Daniel H. S. Leal, Elaine F. F. da Cunha, Kamil Kuca, and Teodorico C. Ramalho. 2019. "Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments" International Journal of Molecular Sciences 20, no. 8: 1829. https://doi.org/10.3390/ijms20081829
APA StyleSales, T. A., Prandi, I. G., de Castro, A. A., Leal, D. H. S., da Cunha, E. F. F., Kuca, K., & Ramalho, T. C. (2019). Recent Developments in Metal-Based Drugs and Chelating Agents for Neurodegenerative Diseases Treatments. International Journal of Molecular Sciences, 20(8), 1829. https://doi.org/10.3390/ijms20081829