Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases



Abstract
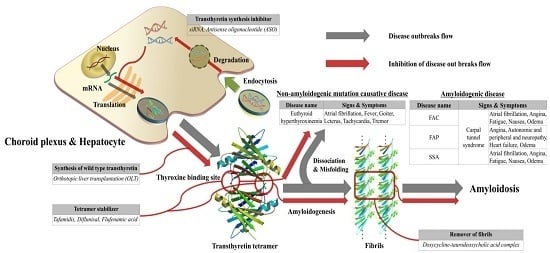
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
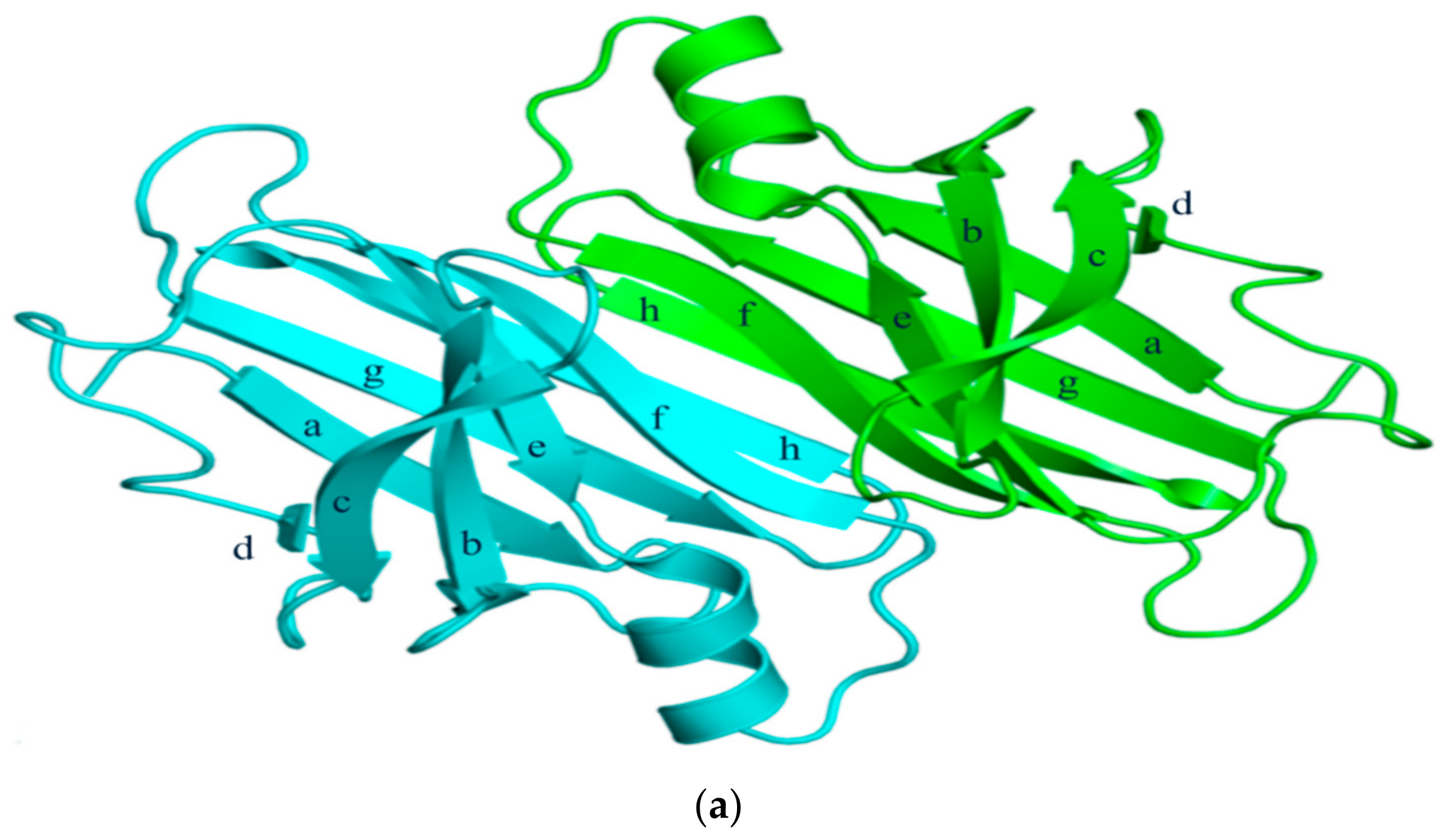
2. Structural Overview: The Role of TTR
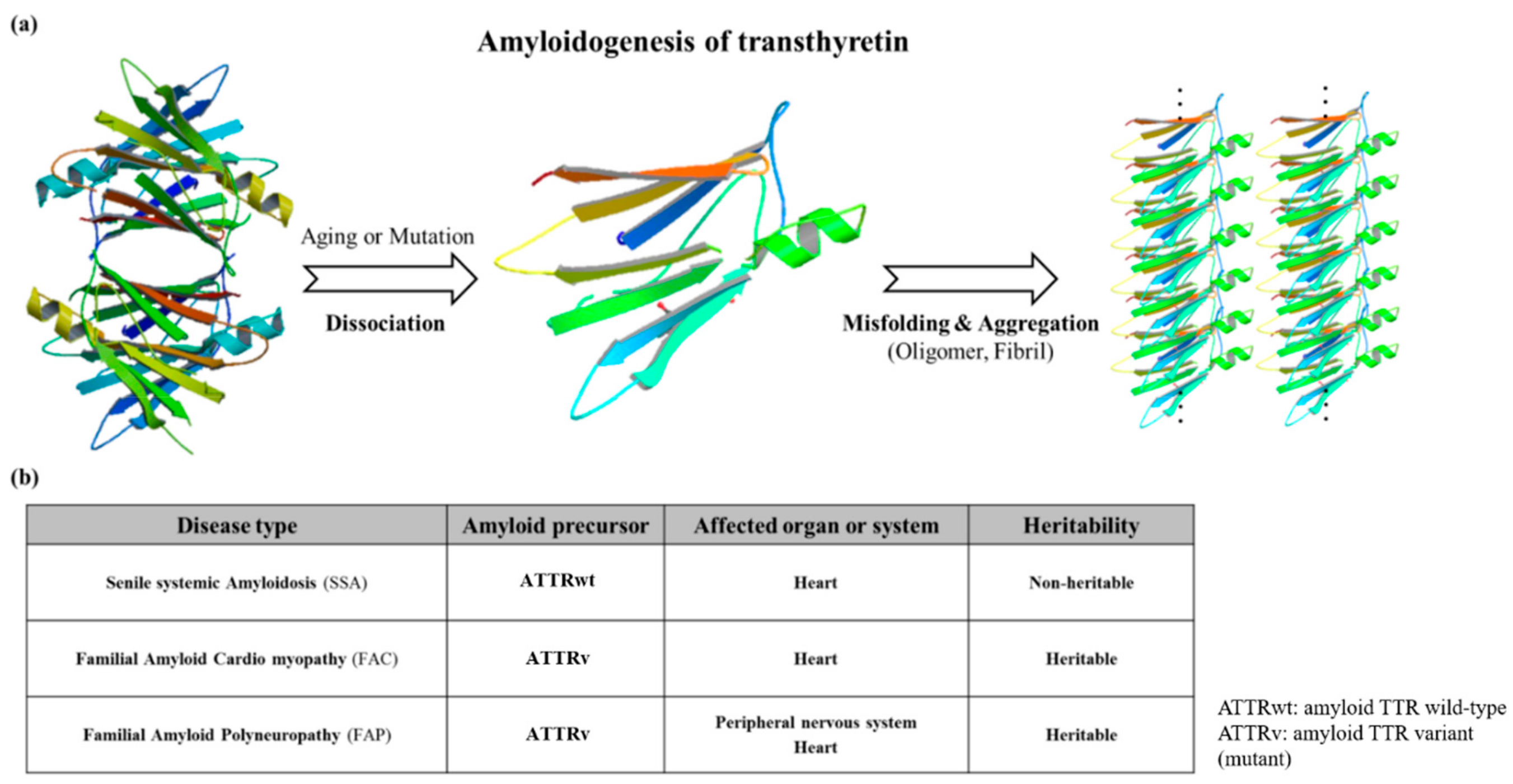
3. ATTR Causative Diseases
4. General Diagnostic Workflow for ATTR Causative Diseases
5. Role of ATTR as Biomarker for Amyloidosis
6. Treatment Methods of TTR Amyloidosis
7. Conclusion and Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kabat, E.A.; Moore, D.H.; Landow, H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J. Clin. Investig. 1942, 21, 571. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.R.; Naylor, S.L.; Kluve-Beckerman, B.; Long, G.L.; McDonald, L.; Shows, T.B.; Benson, M.D. Localization of the human prealbumin gene to chromosome 18. Biochem. Biophys. Res. Commun. 1985, 129, 753–758. [Google Scholar] [CrossRef]
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat. 2014, 35, E2403–E2412. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Trovato, A.; Seno, F.; Folli, C.; Alfieri, B.; Zanotti, G.; Berni, R. Amyloidogenic Potential of Transthyretin Variants: Insights from structural and computational analyses. J. Biol. Chem. 2009, 284, 25832–25841. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.L.; Logue, M.W.; Chan, G.G.; Spencer, B.H.; Prokaeva, T.B.; Baldwin, C.T.; Seldin, D.C.; Connors, L.H. Genetic variation of the transthyretin gene in wild-type transthyretin amyloidosis (ATTRwt). Hum. Genet. 2015, 134, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Lahuerta Pueyo, C.; Aibar Arregui, M.Á.; Gracia Gutierrez, A.; Bueno Juana, E.; Menao Guillén, S. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur. J. Hum. Genet. 2019, 27, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Conti, S.; Mannini, B.; Li, X.; Buxbaum, J.N.; Tiribilli, B.; Chiti, F.; Cecchi, C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim. Et Biophys. Acta (Bba)—Mol. Basis Dis. 2013, 1832, 2302–2314. [Google Scholar] [CrossRef]
- Yang, D.T.; Joshi, G.; Cho, P.Y.; Johnson, J.A.; Murphy, R.M. Transthyretin as both a sensor and a scavenger of β-amyloid oligomers. Biochemistry 2013, 52, 2849–2861. [Google Scholar] [CrossRef]
- Gimeno, A.; Santos, L.M.; Alemi, M.; Rivas, J.; Blasi, D.; Cotrina, E.Y.; Llop, J.; Valencia, G.; Cardoso, I.; Quintana, J.; et al. Insights on the Interaction between Transthyretin and Aβ in Solution. A Saturation Transfer Difference (STD) NMR Analysis of the Role of Iododiflunisal. J. Med. Chem. 2017, 60, 5749–5758. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of Transthyretin Inhibition of β-Amyloid Aggregation in vitro. J. Neurosci. 2013, 33, 19423. [Google Scholar] [CrossRef]
- Xiang, Q.; Bi, R.; Xu, M.; Zhang, D.F.; Tan, L.; Zhang, C.; Fang, Y.; Yao, Y.G. Rare Genetic Variants of the Transthyretin Gene Are Associated with Alzheimer’s Disease in Han Chinese. Mol. Neurobiol. 2017, 54, 5192–5200. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.F.; Palomaki, G.E.; Neveux, L.M.; Navolotskaia, O.; Ledue, T.B.; Craig, W.Y. Reference distributions for the negative acute-phase serum proteins, albumin, transferrin and transthyretin: A practical, simple and clinically relevant approach in a large cohort. J. Clin. Lab. Anal. 1999, 13, 273–279. [Google Scholar] [CrossRef]
- Marchi, N.; Fazio, V.; Cucullo, L.; Kight, K.; Masaryk, T.; Barnett, G.; Volgelbaum, M.; Kinter, M.; Rasmussen, P.; Mayberg, M.R. Serum transthyretin monomer as a possible marker of blood-to-CSF barrier disruption. J. Neurosci. 2003, 23, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Zheng, Q.-C.; Zhang, J.-L.; Cui, Y.-L.; Chu, W.-T.; Zhang, H.-X. Mutation and low pH effect on the stability as well as unfolding kinetics of transthyretin dimer. Biophys. Chem. 2014, 189, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Mizuguchi, M.; Nabeshima, Y.; Kusaka, K.; Yamada, T.; Hosoya, T.; Ohhara, T.; Kurihara, K.; Tomoyori, K.; Tanaka, I. Hydrogen-bond network and pH sensitivity in transthyretin: Neutron crystal structure of human transthyretin. J. Struct. Biol. 2012, 177, 283–290. [Google Scholar] [CrossRef]
- Blake, C.; Geisow, M.; Oatley, S.; Rerat, B.; Rerat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef]
- Prapunpoj, P.; Leelawatwattana, L. Evolutionary changes to transthyretin: Structure–function relationships. Febs J. 2009, 276, 5330–5341. [Google Scholar] [CrossRef]
- Peterson, S.A.; Klabunde, T.; Lashuel, H.A.; Purkey, H.; Sacchettini, J.C.; Kelly, J.W. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1998, 95, 12956–12960. [Google Scholar] [CrossRef]
- Liz, M.A.; Mar, F.M.; Franquinho, F.; Sousa, M.M. Aboard transthyretin: From transport to cleavage. IUBMB Life 2010, 62, 429–435. [Google Scholar] [CrossRef]
- Ferguson, R.N.; Edelhoch, H.; Saroff, H.A.; Robbins, J.; Cahnmann, H.J. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Biochemistry 1975, 14, 282–289. [Google Scholar] [CrossRef]
- Wojtczak, A.; Cody, V.; Luft, J.R.; Pangborn, W. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-L-thyronine at 2.2 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Melhus, H.; Bavik, C.-O.; Rask, L.; Peterson, P.A.; Eriksson, U. Epitope mapping of a monoclonal antibody that blocks the binding of retinol-binding protein to its receptor. Biochem. Biophys. Res. Commun. 1995, 210, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, M.E.; Ong, D.E. Retinol Binding Protein and Its Interaction with Transthyretin; Landes Bioscience: Austin, TX, USA, 2000. [Google Scholar]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.H.; Goodman, D.S. Vitamin A and retinoids in health and disease. New Engl. J. Med. 1984, 310, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. Multiple functions of vitamin A. Physiol. Rev. 1984, 64, 873–937. [Google Scholar] [CrossRef] [PubMed]
- Noy, N.; Slosberg, E.; Scarlata, S. Interactions of retinol with binding proteins: Studies with retinol-binding protein and with transthyretin. Biochemistry 1992, 31, 11118–11124. [Google Scholar] [CrossRef]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.M.; Brito, R.M.M. Tetramer Dissociation and Monomer Partial Unfolding Precedes Protofibril Formation in Amyloidogenic Transthyretin Variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Judge, D.P.; Maurer, M.S. Familial Amyloid Cardiomyopathy Due to TTR Mutations: An underground Cause of Restrictive Cardiomyopathy. J. Card. Fail. 2009, 15, 464. [Google Scholar] [CrossRef]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845. [Google Scholar] [CrossRef]
- Cornwell, G.G.; Sletten, K.; Johansson, B.; Westermark, P. Evidence that the amyloid fibril protein in senile systemic amyloidosis is derived from normal prealbumin. Biochem. Biophys. Res. Commun. 1988, 154, 648–653. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Connors, S. Senile systemic amyloidosis presenting with heart failure. Arch. Intern. Med. 2005, 165, 1425–1429. [Google Scholar]
- Mohty, D.; Damy, T.; Cosnay, P.; Echahidi, N.; Casset-Senon, D.; Virot, P.; Jaccard, A. Cardiac amyloidosis: Updates in diagnosis and management. Arch. Cardiovasc. Dis. 2013, 106, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.; Al-Sergani, H.; Mourad, W.; Tabbaa, R. Amyloid heart disease: New frontiers and insights in pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2005, 32, 178. [Google Scholar] [PubMed]
- Dubrey, S.W.; Cha, K.; Simms, R.W.; Skinner, M.; Falk, R.H. Electrocardiography and Doppler echocardiography in secondary (AA) amyloidosis. Am. J. Cardiol. 1996, 77, 313–315. [Google Scholar] [CrossRef]
- Drexler, M., Helmut; Coats, M.; Andrew, J.S. Explaining fatigue in congestive heart failure. Annu. Rev. Med. 1996, 47, 241–256. [Google Scholar] [CrossRef]
- McKee, P.A.; Castelli, W.P.; McNamara, P.M.; Kannel, W.B. The natural history of congestive heart failure: The Framingham study. New Engl. J. Med. 1971, 285, 1441–1446. [Google Scholar] [CrossRef]
- Ng, B.; Connors, L.H.; Davidoff, R.; Skinner, M.; Falk, R.H. Senile systemic amyloidosis presenting with heart failure: A comparison with light chain–associated amyloidosis. Arch. Intern. Med. 2005, 165, 1425–1429. [Google Scholar] [CrossRef]
- Redondo, C.; Damas, A.M.; Olofsson, A.; Lundgren, E.; Saraiva, M.J.M. Search for intermediate structures in transthyretin fibrillogenesis: Soluble tetrameric Tyr78Phe TTR expresses a specific epitope present only in amyloid fibrils. J. Mol. Biol. 2000, 304, 461–470. [Google Scholar] [CrossRef]
- Andrade, C. A peculiar form of peripheral neuropathy. Brain 1952, 75, 408–427. [Google Scholar] [CrossRef]
- Akiya, S.; Nishio, Y.; Ibi, K.; Uozumi, H.; Takahashi, H.; Hamada, T.; Onishi, A.; Ishiguchi, H.; Hoshii, Y.; Nakazato, M. Lattice corneal dystrophy type II associated with familial amyloid polyneuropathy type IV. Ophthalmology 1996, 103, 1106–1110. [Google Scholar] [CrossRef]
- Steiner, R.D.; Evans, J.P.; Paunio, T.; Uemichi, T.; Benson, M.D. Asp187Asn mutation of gelsolin in an American kindred with familial amyloidosis, Finnish type (FAP IV). Hum. Genet. 1995, 95, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, H.; Ando, Y.; Makita, Y.; Onouchi, Y.; Nakajima, T.; Saraiva, M.; Terazaki, H.; Suhr, O.; Sobue, G.; Nakamura, M. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J. Med Genet. 2004, 41, e51. [Google Scholar] [CrossRef] [PubMed]
- Zaros, C.; Genin, E.; Hellman, U.; Saporta, M.; Languille, L.; Wadington-Cruz, M.; Suhr, O.; Misrahi, M.; Planté-Bordeneuve, V. On the origin of the transthyretin Val30Met familial amyloid polyneuropathy. Ann. Hum. Genet. 2008, 72, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Hund, E.; Linke, R.; Willig, F.; Grau, A. Transthyretin-associated neuropathic amyloidosis pathogenesis and treatment. Neurology 2001, 56, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Wixner, J.; Anan, I.; Lundgren, H.-E.; Wijayatunga, P.; Westermark, P.; Ihse, E. Amyloid fibril composition within hereditary Val30Met (p. Val50Met) transthyretin amyloidosis families. PLoS ONE 2019, 14, e0211983. [Google Scholar] [CrossRef]
- Koike, H.; Misu, K.-I.; Ikeda, S.-I.; Ando, Y.; Nakazato, M.; Ando, E.; Yamamoto, M.; Hattori, N.; Sobue, G.; Japan, F.T.S.G.F.H.N.I. Type I (Transthyretin Met30) Familial Amyloid Polyneuropathy in Japan: Early- vs. Late-Onset Form. JAMA Neurol. 2002, 59, 1771–1776. [Google Scholar] [CrossRef]
- Andreou, S.; Panayiotou, E.; Michailidou, K.; Pirpa, P.; Hadjisavvas, A.; El Salloukh, A.; Barnes, D.; Antoniou, A.; Agathangelou, P.; Papastavrou, K.; et al. Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset. Amyloid 2018, 25, 220–226. [Google Scholar] [CrossRef]
- Conceição, I.; De Carvalho, M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): Comparison between late- and early-onset cases in Portugal. Muscle Nerve 2007, 35, 116–118. [Google Scholar] [CrossRef]
- Pinto, M.V.; Pinto, L.F.; Dias, M.; Rosa, R.S.; Mundayat, R.; Pedrosa, R.C.; Waddington-Cruz, M. Late-onset hereditary ATTR V30M amyloidosis with polyneuropathy: Characterization of Brazilian subjects from the THAOS registry. J. Neurol. Sci. 2019, 403, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ando, Y.; Ueda, M.; Kawagashira, Y.; Iijima, M.; Fujitake, J.; Hayashi, M.; Yamamoto, M.; Mukai, E.; Nakamura, T.; et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J. Neurol. Sci. 2009, 287, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.R.; Conneally, P.M.; Benson, M.D. A DNA test for Indiana/Swiss hereditary amyloidosis (FAP II). Am. J. Hum. Genet. 1988, 43, 182–187. [Google Scholar] [PubMed]
- Sekijima, Y.; Yoshida, K.; Tokuda, T.; Ikeda, S.-I. Familial Transthyretin Amyloidosis; U.S. Department of Health & Human Services: Washington, DC, USA, 2018. Available online: https://rarediseases.info.nih.gov/diseases/656/familial-transthyretin-amyloidosis.
- Koga, T.; Ando, E.; Hirata, A.; Fukushima, M.; Kimura, A.; Ando, Y.; Negi, A.; Tanihara, H. Vitreous opacities and outcome of vitreous surgery in patients with familial amyloidotic polyneuropathy. Am. J. Ophthalmol. 2003, 135, 188–193. [Google Scholar] [CrossRef]
- Misu, K.-i.; Hattori, N.; Nagamatsu, M.; Ikeda, S.-I.; Ando, Y.; Nakazato, M.; Takei, Y.-i.; Hanyu, N.; Usui, Y.; Tanaka, F. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Brain 1999, 122, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- OLOFSSON, B.O.; Grankvist, K.; Boman, K.; Forsberg, K.; Lafvas, I.; Lithner, F. Assessment of thyroid and adrenal function in patients with familial amyloidotic polyneuropathy. J. Intern. Med. 1989, 225, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Connection, B.H. Contemporary Reviews in Cardiovascular Medicine. Circulation 2007, 116, 77–84. [Google Scholar]
- Ranløv, I.; Alves, I.L.; Ranløv, P.J.; Husby, G.; Costa, P.P.; Saraiva, M.J. A Danish kindred with familial amyloid cardiomyopathy revisited: Identification of a mutant transthyretinmethionine 111 variant in serum from patients and carriers. Am. J. Med. 1992, 93, 3–8. [Google Scholar] [CrossRef]
- Jacobson, D.R.; Pastore, R.D.; Yaghoubian, R.; Kane, I.; Gallo, G.; Buck, F.S.; Buxbaum, J.N. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. New Engl. J. Med. 1997, 336, 466–473. [Google Scholar] [CrossRef]
- Sattianayagam, P.T.; Hahn, A.F.; Whelan, C.J.; Gibbs, S.D.; Pinney, J.H.; Stangou, A.J.; Rowczenio, D.; Pflugfelder, P.W.; Fox, Z.; Lachmann, H.J. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur. Heart J. 2012, 33, 1120–1127. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Maurer, M.S.; Judge, D.P.; Zeldenrust, S.; Skinner, M.; Kim, A.Y.; Falk, R.H.; Cheung, K.N.; Patel, A.R.; Pano, A. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am. Heart J. 2012, 164, 222–228.e221. [Google Scholar] [CrossRef] [PubMed]
- Polimanti, R.; Nuñez, Y.Z.; Gelernter, J. Increased Risk of Multiple Outpatient Surgeries in African-American Carriers of Transthyretin Val122Ile Mutation Is Modulated by Non-Coding Variants. J. Clin. Med. 2019, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.; Jacobson, D.R.; Tagoe, C.; Alexander, A.; Kitzman, D.W.; Greenberg, B.; Thaneemit-Chen, S.; Lavori, P. Transthyretin V122I in African Americans with congestive heart failure. J. Am. Coll. Cardiol. 2006, 47, 1724–1725. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.-I.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet. J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J.M. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum. Mutat. 2001, 17, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Alves, I.L.; Saraiva, M.J.; Thibodeau, S.N.; Buxbaum, J.N. Transthyretin Ser 6 gene frequency in individuals without amyloidosis. Hum. Genet. 1995, 95, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Refetoff, S.; Marinov, V.; Tunca, H.; Byrne, M.M.; Sunthornthepvarakul, T.; Weiss, R. A new family with hyperthyroxinemia caused by transthyretin Val109 misdiagnosed as thyrotoxicosis and resistance to thyroid hormone--a clinical research center study. J. Clin. Endocrinol. Metab. 1996, 81, 3335–3340. [Google Scholar] [PubMed]
- Banypersad, S.M.; Moon, J.C.; Whelan, C.; Hawkins, P.N.; Wechalekar, A.D. Updates in cardiac amyloidosis: A review. J. Am. Heart Assoc. 2012, 1, e000364. [Google Scholar] [CrossRef]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef]
- Maceira, A.M.; Joshi, J.; Prasad, S.K.; Moon, J.C.; Perugini, E.; Harding, I.; Sheppard, M.N.; Poole-Wilson, P.A.; Hawkins, P.N.; Pennell, D.J. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005, 111, 186–193. [Google Scholar] [CrossRef]
- MEYER, R.A.; KAPLAN, S. Echocardiography in the Diagnosis of Hypoplasia of the Left or Right Ventricles in the Neonate. Circulation 1972, 46, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tissot, C.; Singh, Y.; Sekarski, N. Echocardiographic Evaluation of Ventricular Function—For the Neonatologist and Pediatric Intensivist. Front. Pediatrics 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Jurcut, R.; Giusca, S.; La Gerche, A.; Vasile, S.; Ginghina, C.; Voigt, J.-U. The echocardiographic assessment of the right ventricle: What to do in 2010? Eur. Heart J. Cardiovasc. Imaging 2010, 11, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.M.; Arushi, A.; Stapleton, D.; Reddy, P.C. Assessment of right ventricle by echocardiogram. In Echocardiography in Heart Failure and Cardiac Electrophysiology; Lakshmanadoss, U., Ed.; IntechOpen Limited: London, UK, 19 October 2016. [Google Scholar]
- Quinones, M.A.; Gaasch, W.H.; Alexander, J.K. Echocardiographic assessment of left ventricular function with special reference to normalized velocities. Circulation 1974, 50, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Potter, E.; Marwick, T.H. Assessment of Left Ventricular Function by Echocardiography: The Case for Routinely Adding Global Longitudinal Strain to Ejection Fraction. JACC: Cardiovasc. Imaging 2018, 11, 260–274. [Google Scholar]
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimaraes, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: Evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 2001, 159, 1993–2000. [Google Scholar] [CrossRef]
- Ihse, E.; Ybo, A.; Suhr, O.B.; Lindqvist, P.; Backman, C.; Westermark, P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J. Pathol. 2008, 216, 253–261. [Google Scholar] [CrossRef]
- Jamet, M.-P.; Gnemmi, V.; Hachulla, É.; Dhaenens, C.-M.; Bouchindhomme, B.; Delattre, C.; Glowacki, F.; Hatron, P.-Y.; Lacour, A.; Lamblin, N. Distinctive patterns of transthyretin amyloid in salivary tissue: A clinicopathologic study of 92 patients with amyloid-containing minor salivary gland biopsies. Am. J. Surg. Pathol. 2015, 39, 1035–1044. [Google Scholar] [CrossRef]
- Satoskar, A.A.; Efebera, Y.; Hasan, A.; Brodsky, S.; Nadasdy, G.; Dogan, A.; Nadasdy, T. Strong tranthyretin immunostaining, potential pitfall in cardiac amyloid typing. Am. J. Surg. Pathol. 2011, 35, 1685. [Google Scholar] [CrossRef]
- Ebenezer, G.J.; Liu, Y.; Judge, D.P.; Cunningham, K.; Truelove, S.; Carter, N.D.; Sebastian, B.; Byrnes, K.; Polydefkis, M. Cutaneous nerve biomarkers in transthyretin familial amyloid polyneuropathy. Ann. Neurol. 2017, 82, 44–56. [Google Scholar] [CrossRef]
- Soares, M.L.; Coelho, T.; Sousa, A.; Batalov, S.; Conceição, I.; Sales-Luís, M.L.; Ritchie, M.D.; Williams, S.M.; Nievergelt, C.M.; Schork, N.J. Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: Complexity in a single-gene disease. Hum. Mol. Genet. 2005, 14, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Asl, K.H.; Yazaki, M.; Benson, M.D. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid 2005, 12, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Tojo, K.; Sekijima, Y.; Kelly, J.W.; Ikeda, S.-I. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci. Res. 2006, 56, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, M.; Finch, N.; Baker, M.; Wang, X.; Bieniek, K.; Dejesus-Hernandez, M.; Gendron, T.; Asmann, Y.; Heckman, M.; Brown, P. Transthyretin as Potential Biomarker for C9ORF72-related Diseases (I8-2A). Neurology 2015, 84, I8–2A. [Google Scholar]
- Sharma, M.; Khan, S.; Rahman, S.; Singh, L.R. The Extracellular Protein, Transthyretin Is an Oxidative Stress Biomarker. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Li, X.; Masliah, E.; Reixach, N.; Buxbaum, J.N. Neuronal Production of Transthyretin in Human and Murine Alzheimer’s Disease: Is It Protective? J. Neurosci. 2011, 31, 12483–12490. [Google Scholar] [CrossRef]
- Schonhoft, J.D.; Monteiro, C.; Plate, L.; Eisele, Y.S.; Kelly, J.M.; Boland, D.; Parker, C.G.; Cravatt, B.F.; Teruya, S.; Helmke, S.; et al. Peptide probes detect misfolded transthyretin oligomers in plasma of hereditary amyloidosis patients. Sci. Transl. Med. 2017, 9, eaam7621. [Google Scholar] [CrossRef]
- Stangou, A.; Hawkins, P.; Heaton, N.; Rela, M.; Monaghan, M.; Nihoyannopoulos, P.; O’Grady, J.; Pepys, M.; Williams, R. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: Implications for amyloid fibrillogenesis. Transplantation 1998, 66, 229–233. [Google Scholar] [CrossRef]
- Holmgren, G.; Steen, L.; Ekstedt, J.; Groth, C.G.; Ericzon, B.G.; Eriksson, S.; Andersen, O.; Karlberg, I.; Nordén, G.; Nakazato, M. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin. Genet. 1991, 40, 242–246. [Google Scholar] [CrossRef]
- Rocha, A.; Lobato, L.; Silva, H.; Beirao, I.; Santos, J.; Pessegueiro, H.; Almeida, R.; Cabrita, A. Characterization of end-stage renal disease after liver transplantation in transthyretin amyloidosis (ATTR V30M). Transplant. Proc. 2011, 43, 189–193. [Google Scholar] [CrossRef]
- Hamour, I.; Lachmann, H.; Goodman, H.; Petrou, M.; Burke, M.; Hawkins, P.; Banner, N. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am. J. Transplant. 2008, 8, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.F.; Carithers, R.L. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology 2005, 41, 1407–1432. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M.; Michl, K. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): Developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005, 112, e154–e235. [Google Scholar] [PubMed]
- Benson, M.D.; Kluve-Beckerman, B.; Zeldenrust, S.R.; Siesky, A.M.; Bodenmiller, D.M.; Showalter, A.D.; Sloop, K.W. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve 2006, 33, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, E.J.; Guo, S.; Booten, S.; Alvarado, L.; Benson, M.; Hughes, S.; Monia, B.P. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid 2012, 19, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef]
- Adams, D. Recent advances in the treatment of familial amyloid neuropathies. Ther. Adv. Neurol. Disord. 2012, 1756285612470192. [Google Scholar]
- Benson, M.D.; Dasgupta, N.R.; Monia, B.P. Inotersen (transthyretin-specific antisense oligonucleotide) for treatment of transthyretin amyloidosis. Neurodegener. Dis. Manag. 2018, 9, 25–30. [Google Scholar] [CrossRef]
- Wood, H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 2018, 14, 570. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2018, 9, 5–23. [Google Scholar] [CrossRef]
- Yang, J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev. Clin. Pharmacol. 2019, 12, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. New Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Baures, P.W.; Oza, V.B.; Peterson, S.A.; Kelly, J.W. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorganic Med. Chem. 1999, 7, 1339–1347. [Google Scholar] [CrossRef]
- Dubrey, S.W.; Dubrey, S. An update on treatments for amyloid heart disease. Br. J. Cardiol. 2013, 20, 107–109. [Google Scholar]
- Berk, J.; Dyck, P.; Obici, L.; Zeldenrust, S.; Sekijima, Y.; Yamashita, T.; Ando, Y.; Ikeda, S.-I.; Gorevic, P.; Merlini, G. The diflunisal trial: Update on study drug tolerance and disease progression. Amyloid 2011, 18, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Galant, N.J.; Bugyei-Twum, A.; Rakhit, R.; Walsh, P.; Sharpe, S.; Arslan, P.E.; Westermark, P.; Higaki, J.N.; Torres, R.; Tapia, J.; et al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: A potential diagnostic and therapeutic for TTR amyloidoses. Sci. Rep. 2016, 6, 25080. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, I.; Martins, D.; Ribeiro, T.; Merlini, G.; Saraiva, M.J. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: Studies in FAP mouse models. J. Transl. Med. 2010, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Haupt, M.; Blakeley, M.P.; Fisher, S.J.; Mason, S.A.; Cooper, J.B.; Mitchell, E.P.; Forsyth, V.T. Binding site asymmetry in human transthyretin: Insights from a joint neutron and X-ray crystallographic analysis using perdeuterated protein. IUCrJ 2014, 1, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.C.; Freire, J.B.B.; Palhano, F.L.; Azevedo, E.P.C.; Foguel, D.; Lima, L.M.T.R. Crystal structure of human transthyretin variant A25T—#1, in press.






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, G.Y.; Jamerlan, A.; Shim, K.H.; An, S.S.A. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int. J. Mol. Sci. 2019, 20, 2982. https://doi.org/10.3390/ijms20122982
Park GY, Jamerlan A, Shim KH, An SSA. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. International Journal of Molecular Sciences. 2019; 20(12):2982. https://doi.org/10.3390/ijms20122982
Chicago/Turabian StylePark, Gil Yong, Angelo Jamerlan, Kyu Hwan Shim, and Seong Soo A. An. 2019. "Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases" International Journal of Molecular Sciences 20, no. 12: 2982. https://doi.org/10.3390/ijms20122982
APA StylePark, G. Y., Jamerlan, A., Shim, K. H., & An, S. S. A. (2019). Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. International Journal of Molecular Sciences, 20(12), 2982. https://doi.org/10.3390/ijms20122982