Multigenerational and Transgenerational Effects of Dioxins


Abstract
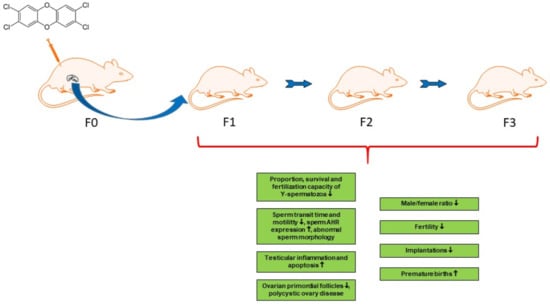
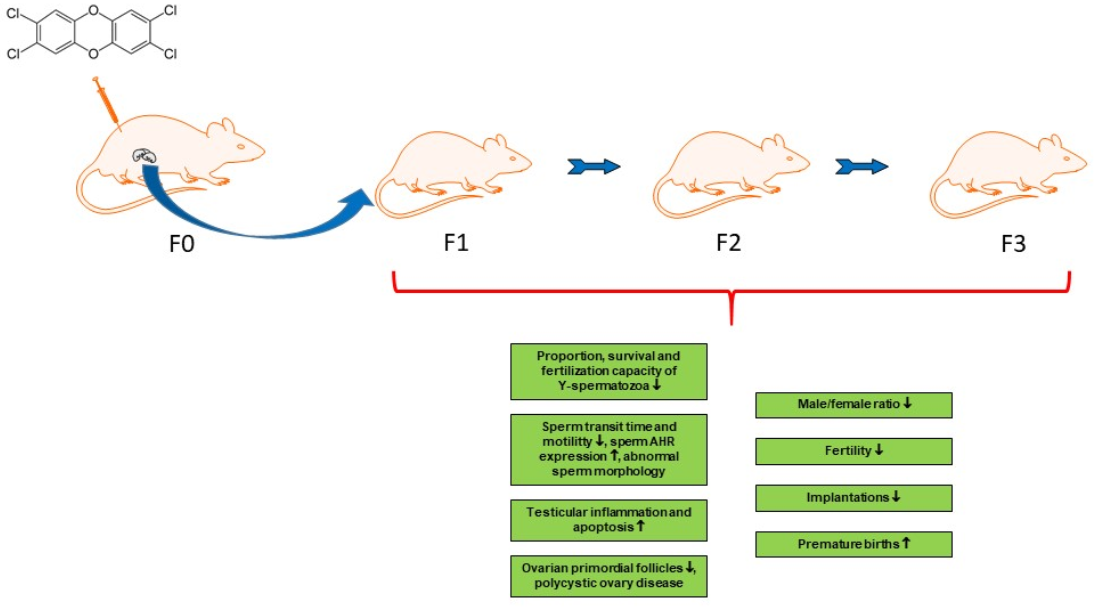
1. Introduction
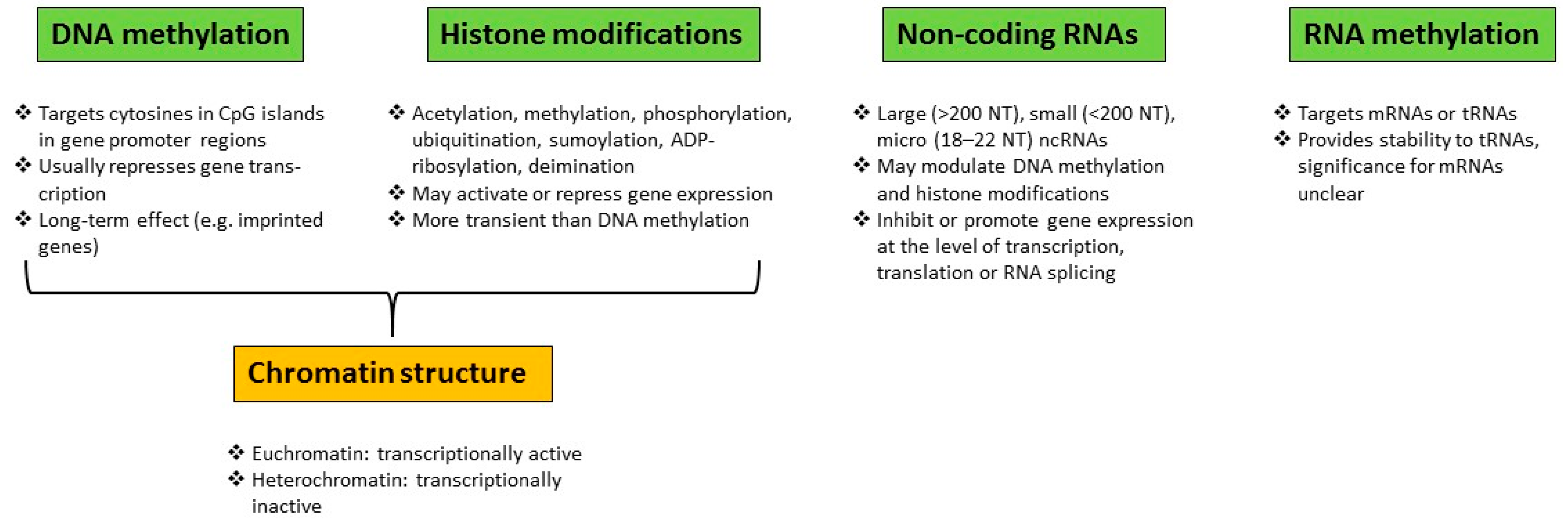
1.1. Epigenetic Alterations and Environmental Factors
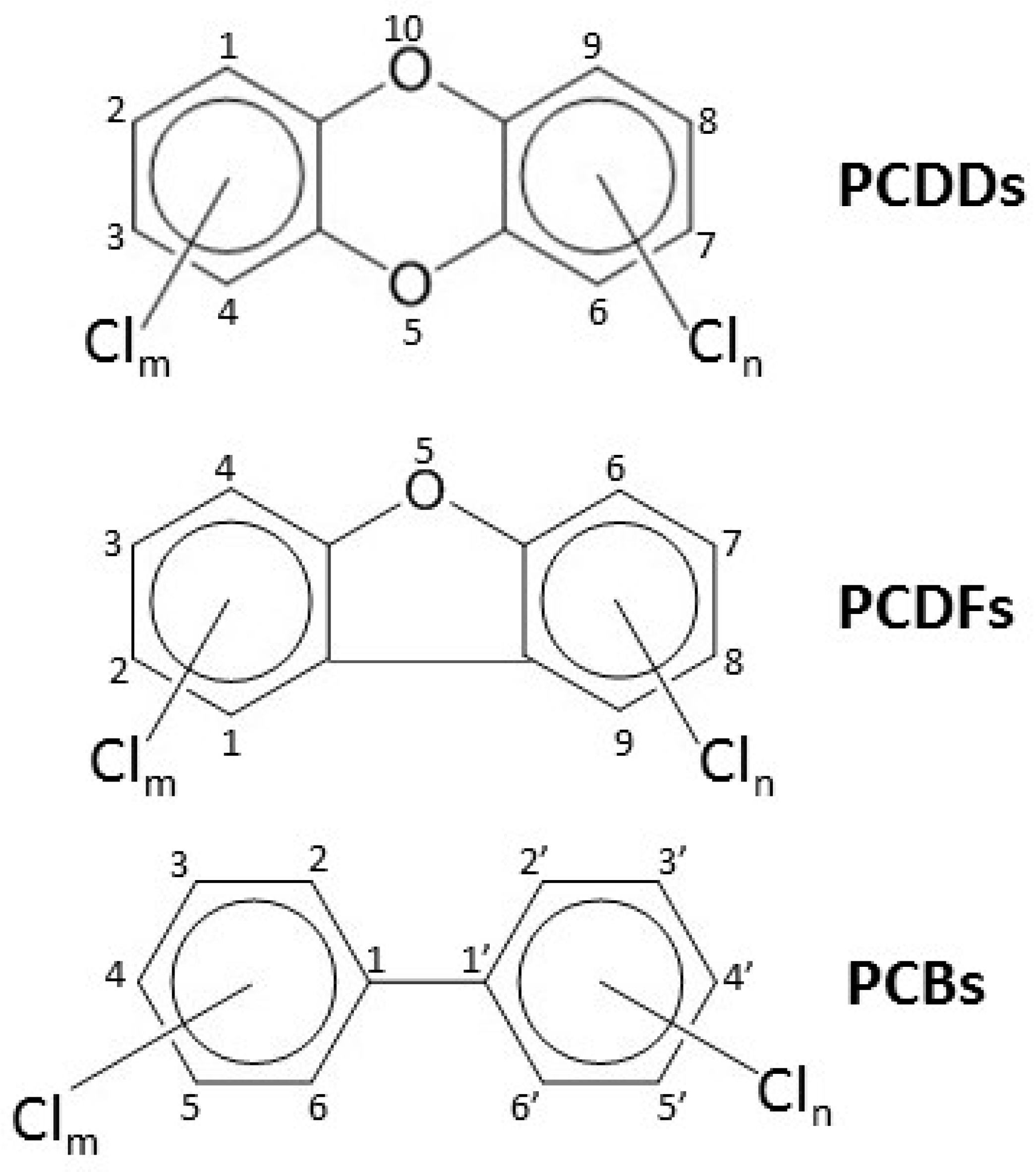
1.2. Dioxins
1.3. Aryl Hydrocarbon Receptor (AHR) and the Molecular Mechanism of Action of Dioxins
1.3.1. Canonical Pathway
1.3.2. Non-Canonical Pathway
Epigenetic Modifications
2. Paternally or Maternally Mediated Effects on the Next Generation
2.1. Effects on Male/Female Sex Ratio
2.1.1. Humans
2.1.2. Laboratory Animals
2.2. Effects on Pregnancy Outcome
3. Paternally or Maternally Mediated Multigenerational and Transgenerational Effects
4. Paternally and Maternally Mediated Multigenerational and Transgenerational Effects
5. Conclusions and Future Prospects
Funding
Conflicts of Interest
References
- Schug, T.T.; Barouki, R.; Gluckman, P.D.; Grandjean, P.; Hanson, M.; Heindel, J.J. PPTOX III: Environmental stressors in the developmental origins of disease—Evidence and mechanisms. Toxicol. Sci. 2013, 131, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Gluckman, P.D. Early developmental conditioning of later health and disease: Physiology or pathophysiology? Physiol. Rev. 2014, 94, 1027–1076. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A. Epigenetics as a Driver of Developmental Origins of Health and Disease: Did We Forget the Fathers? Bioessays 2018, 40. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A. POHaD: Why we should study future fathers. Environ. Epigenet. 2018, 4, dvy007. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.E.; Sadler-Riggleman, I.; Skinner, M.K. Environmentally induced epigenetic transgenerational inheritance of disease. Environ. Epigenet. 2018, 4, dvy016. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Bruschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; et al. Risk for animal and human health related to the presence of dioxins and dioxin-like PCBs in feed and food. EFSA J. 2018, 16, 5333. [Google Scholar]
- Pilsner, J.R.; Parker, M.; Sergeyev, O.; Suvorov, A. Spermatogenesis disruption by dioxins: Epigenetic reprograming and windows of susceptibility. Reprod. Toxicol. 2017, 69, 221–229. [Google Scholar] [CrossRef]
- Brehm, E.; Flaws, J.A. Transgenerational effects of endocrine disrupting chemicals on male and female reproduction. Endocrinology 2019, 160, 1421–1435. [Google Scholar] [CrossRef]
- Hanna, C.W.; Kelsey, G. The specification of imprints in mammals. Heredity 2014, 113, 176–183. [Google Scholar] [CrossRef]
- Skinner, M.K. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod. Toxicol. 2008, 25, 2–6. [Google Scholar] [CrossRef]
- Skinner, M.K. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell. Endocrinol. 2014, 398, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, E.L.; Jacobs, M.N.; Gant, T.W. Environmentally induced epigenetic toxicity: Potential public health concerns. Crit. Rev. Toxicol. 2016, 46, 676–700. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.R.; Veselovska, L.; Kelsey, G. Establishment and functions of DNA methylation in the germline. Epigenomics 2016, 8, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Kimmig, J.; Schulz, K.H. Chlorierte aromatische Zyklische Äther als Ursache der sogenannten Chlorakne. Natürwissenschaften 1957, 44, 337–338. [Google Scholar] [CrossRef]
- Niwa, A.; Kumaki, K.; Nebert, D.W.; Poland, A.P. Genetic expression of aryl hydrocarbon hydroxylase activity in the mouse. Distinction between the "responsive" homozygote and heterozygote at the Ah locus. Arch. Biochem. Biophys. 1975, 166, 559–564. [Google Scholar] [CrossRef]
- Atlas, A.S.; Thorgeirsson, S.S.; Boobis, A.R.; Kumaki, K.; Nebert, D.W. Differential induction of murine Ah locus-associated monooxygenase activities in rabbit liver and kidney. Biochem. Pharmacol. 1975, 24, 2111–2116. [Google Scholar] [CrossRef]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar]
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 1992, 184, 246–253. [Google Scholar] [CrossRef]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA. 1992, 89, 8185–8189. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G. Role of chaperone proteins in AHR function. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 47–61. [Google Scholar]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. The immunophilin-like protein XAP2 regulates ubiquitination and subcellular localization of the dioxin receptor. J. Biol. Chem. 2000, 275, 41317–41324. [Google Scholar] [CrossRef]
- Kazlauskas, A.; Sundstrom, S.; Poellinger, L.; Pongratz, I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol. Cell. Biol. 2001, 21, 2594–2607. [Google Scholar] [CrossRef] [PubMed]
- Henry, E.C.; Gasiewicz, T.A. Agonist but not antagonist ligands induce conformational change in the mouse aryl hydrocarbon receptor as detected by partial proteolysis. Mol. Pharmacol. 2003, 63, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.; Denison, M.S. Role of the Per/Arnt/Sim domains in ligand-dependent transformation of the aryl hydrocarbon receptor. J. Biol. Chem. 2008, 283, 32995–33005. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.A.; Tillitt, D.E.; Hannink, M. Regulation of subcellular localization of the aryl hydrocarbon receptor (AhR). Arch. Biochem. Biophys. 2001, 389, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Swanson, H. Dioxin response elements and regulation of gene transcription. In AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 81–91. [Google Scholar]
- Okino, S.T.; Whitlock, J.P., Jr. Dioxin induces localized, graded changes in chromatin structure: Implications for Cyp1A1 gene transcription. Mol. Cell. Biol. 1995, 15, 3714–3721. [Google Scholar] [CrossRef]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2005, 433, 379–386. [Google Scholar] [CrossRef]
- Ikuta, T.; Eguchi, H.; Tachibana, T.; Yoneda, Y.; Kawajiri, K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 1998, 273, 2895–2904. [Google Scholar] [CrossRef]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1999, 274, 28708–28715. [Google Scholar] [CrossRef]
- Roberts, B.J.; Whitelaw, M.L. Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. J. Biol. Chem. 1999, 274, 36351–36356. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Baldwin, K.T. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activation and DNA binding of AhR. J. Biol. Chem. 2000, 275, 8432–8438. [Google Scholar] [CrossRef] [PubMed]
- Arpiainen, S.; Raffalli-Mathieu, F.; Lang, M.A.; Pelkonen, O.; Hakkola, J. Regulation of the Cyp2a5 gene involves an aryl hydrocarbon receptor-dependent pathway. Mol. Pharmacol. 2005, 67, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Bandiera, S.M. Characterization of a new cytochrome P450 enzyme, CYP2S1, in rats: Its regulation by aryl hydrocarbon receptor agonists. Toxicology 2010, 267, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, S.; Ehmer, U.; Freiberg, N.; Manns, M.P.; Strassburg, C.P. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyltransferase 1A10. J. Biol. Chem. 2010, 285, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Renaud, H.J.; Klaassen, C.D.; Cui, J.Y. Age-Specific Regulation of Drug-Processing Genes in Mouse Liver by Ligands of Xenobiotic-Sensing Transcription Factors. Drug Metab. Dispos. 2016, 44, 1038–1049. [Google Scholar] [CrossRef]
- Ma, Q. Overview of AHR functional domains and the classical AHR signaling pathway: Induction of drug metabolizing enzymes. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 35–45. [Google Scholar]
- Yeager, R.L.; Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 2009, 111, 238–246. [Google Scholar] [CrossRef]
- Boverhof, D.R.; Burgoon, L.D.; Tashiro, C.; Sharratt, B.; Chittim, B.; Harkema, J.R.; Mendrick, D.L.; Zacharewski, T.R. Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicol. Sci. 2006, 94, 398–416. [Google Scholar] [CrossRef]
- Boutros, P.C.; Yan, R.; Moffat, I.D.; Pohjanvirta, R.; Okey, A.B. Transcriptomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in liver: Comparison of rat and mouse. BMC Genom. 2008, 9, 419. [Google Scholar] [CrossRef]
- Franc, M.A.; Moffat, I.D.; Boutros, P.C.; Tuomisto, J.T.; Tuomisto, J.; Pohjanvirta, R.; Okey, A.B. Patterns of dioxin-altered mRNA expression in livers of dioxin-sensitive versus dioxin-resistant rats. Arch. Toxicol. 2008, 82, 809–830. [Google Scholar] [CrossRef]
- Tijet, N.; Boutros, P.C.; Moffat, I.D.; Okey, A.B.; Tuomisto, J.; Pohjanvirta, R. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 2006, 69, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Riddick, D.S.; Lee, C.; Bhathena, A.; Timsit, Y.E.; Cheng, P.Y.; Morgan, E.T.; Prough, R.A.; Ripp, S.L.; Miller, K.K.; Jahan, A.; et al. Transcriptional suppression of cytochrome P450 genes by endogenous and exogenous chemicals. Drug Metab. Dispos. 2004, 32, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef]
- MacPherson, L.; Tamblyn, L.; Rajendra, S.; Bralha, F.; McPherson, J.P.; Matthews, J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acids Res. 2013, 41, 1604–1621. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, L.; Ahmed, S.; Tamblyn, L.; Krutmann, J.; Forster, I.; Weighardt, H.; Matthews, J. Aryl hydrocarbon receptor repressor and TiPARP (ARTD14) use similar, but also distinct mechanisms to repress aryl hydrocarbon receptor signaling. Int. J. Mol. Sci. 2014, 15, 7939–7957. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol. 2009, 77, 608–626. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, F. Nongenomic route of action of TCDD: Identity, characteristics, and toxicological significance. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 197–215. [Google Scholar]
- Patel, R.D.; Murray, I.A.; Flaveny, C.A.; Kusnadi, A.; Perdew, G.H. Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab. Investig. 2009, 89, 695–707. [Google Scholar] [CrossRef]
- Li, W.; Vogel, C.F.; Wu, D.; Matsumura, F. Non-genomic action of TCDD to induce inflammatory responses in HepG2 human hepatoma cells and in liver of C57BL/6J mice. Biol. Chem. 2010, 391, 1205–1219. [Google Scholar] [CrossRef]
- Tsai, M.J.; Hsu, Y.L.; Wang, T.N.; Wu, L.Y.; Lien, C.T.; Hung, C.H.; Kuo, P.L.; Huang, M.S. Aryl hydrocarbon receptor (AhR) agonists increase airway epithelial matrix metalloproteinase activity. J. Mol. Med. 2014, 92, 615–628. [Google Scholar] [CrossRef]
- Tanos, R.; Patel, R.D.; Murray, I.A.; Smith, P.B.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology 2012, 55, 1994–2004. [Google Scholar] [CrossRef]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Kato, S. The E3 ubiquitin ligase activity of transcription factor AHR permits nongenomic regulation of biological pathways. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 143–156. [Google Scholar]
- Weiss, C.; Faust, D.; Durk, H.; Kolluri, S.K.; Pelzer, A.; Schneider, S.; Dietrich, C.; Oesch, F.; Gottlicher, M. TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38-MAPK-dependent pathway. Oncogene 2005, 24, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, S.K.; Smolenski, A. Phosphodiesterases link the aryl hydrocarbon receptor complex to cyclic nucleotide signaling. Biochem. Pharmacol. 2009, 77, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.H.; Hong, C.H.; Lee, H.Y.; Kang, H.J.; Han, S.W. Enhanced TGF-beta1 is involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induced oxidative stress in C57BL/6 mouse testis. Toxicol. Lett. 2008, 178, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, T.B.; Tomblin, J.K.; Primerano, D.A.; Boskovic, G.; Fan, J.; Mehmi, I.; Fletcher, J.; Santanam, N.; Hurn, E.; Morris, G.Z.; et al. Endogenous aryl hydrocarbon receptor promotes basal and inducible expression of tumor necrosis factor target genes in MCF-7 cancer cells. Biochem. Pharmacol. 2014, 91, 390–399. [Google Scholar] [CrossRef]
- Azkargorta, M.; Fullaondo, A.; Laresgoiti, U.; Aloria, K.; Infante, A.; Arizmendi, J.M.; Zubiaga, A.M. Differential proteomics analysis reveals a role for E2F2 in the regulation of the Ahr pathway in T lymphocytes. Mol. Cell. Proteom. 2010, 9, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Elferink, C.J.; Ge, N.L.; Levine, A. Maximal aryl hydrocarbon receptor activity depends on an interaction with the retinoblastoma protein. Mol. Pharmacol. 2001, 59, 664–673. [Google Scholar] [CrossRef]
- Watabe, T. Roles of old players in the suppression of a new player: Networks for the transcriptional control of angiogenesis. J. Biochem. 2011, 149, 117–119. [Google Scholar] [CrossRef]
- Pääjärvi, G.; Viluksela, M.; Pohjanvirta, R.; Stenius, U.; Högberg, J. TCDD activates Mdm2 and attenuates the p53 response to DNA damaging agents. Carcinogenesis 2005, 26, 201–208. [Google Scholar] [CrossRef]
- Backlund, M.; Ingelman-Sundberg, M. Regulation of aryl hydrocarbon receptor signal transduction by protein tyrosine kinases. Cell. Signal. 2005, 17, 39–48. [Google Scholar] [CrossRef]
- Sutter, C.H.; Yin, H.; Li, Y.; Mammen, J.S.; Bodreddigari, S.; Stevens, G.; Cole, J.A.; Sutter, T.R. EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 4266–4271. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Peng, Z.; Raufman, J.P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1006–G1015. [Google Scholar] [CrossRef] [PubMed]
- Barhoover, M.A.; Hall, J.M.; Greenlee, W.F.; Thomas, R.S. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol. Pharmacol. 2010, 77, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Rabson, A.B.; Germino, J.F.; Gallo, M.A.; Tian, Y. Mechanism of suppression of cytochrome P-450 1A1 expression by tumor necrosis factor-alpha and lipopolysaccharide. J. Biol. Chem. 2001, 276, 39638–39644. [Google Scholar] [CrossRef] [PubMed]
- Braeuning, A.; Kohle, C.; Buchmann, A.; Schwarz, M. Coordinate regulation of cytochrome P450 1a1 expression in mouse liver by the aryl hydrocarbon receptor and the beta-catenin pathway. Toxicol. Sci. 2011, 122, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Pohjanvirta, R.; Spielmann, P.; Viluksela, M.; Buchmann, D.P.; Wenger, R.H.; Gassmann, M. Simultaneous exposure of rats to dioxin and carbon monoxide reduces the xenobiotic but not the hypoxic response. Biol. Chem. 2004, 385, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kleinhenz, D.J.; Rupnow, H.L.; Campbell, A.G.; Thule, P.M.; Sutliff, R.L.; Hart, C.M. The PPARgamma ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul. Pharmacol. 2007, 46, 456–462. [Google Scholar] [CrossRef]
- Shibahara, N.; Masunaga, Y.; Iwano, S.; Yamazaki, H.; Kiyotani, K.; Kamataki, T. Human cytochrome P450 1A1 is a novel target gene of liver X receptor alpha. Drug Metab. Pharmacokinet. 2011, 26, 451–457. [Google Scholar] [CrossRef]
- Brunnberg, S.; Swedenborg, E.; Gustafsson, J.A. Functional interactions of AHR with other receptors. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 127–141. [Google Scholar]
- Tian, Y.; Ke, S.; Thomas, T.; Meeker, R.J.; Gallo, M.A. Transcriptional suppression of estrogen receptor gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. Steroid Biochem. Mol. Biol. 1998, 67, 17–24. [Google Scholar] [CrossRef]
- Spink, D.C.; Hayes, C.L.; Young, N.R.; Christou, M.; Sutter, T.R.; Jefcoate, C.R.; Gierthy, J.F. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on estrogen metabolism in MCF-7 breast cancer cells: Evidence for induction of a novel 17 beta-estradiol 4-hydroxylase. J. Steroid Biochem. Mol. Biol. 1994, 51, 251–258. [Google Scholar] [CrossRef]
- Safe, S.; Wormke, M.; Samudio, I. Mechanisms of inhibitory aryl hydrocarbon receptor-estrogen receptor crosstalk in human breast cancer cells. J. Mammary Gland Biol. Neoplasia 2000, 5, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Wihlen, B.; Thomsen, J.; Gustafsson, J.A. Aryl hydrocarbon receptor-mediated transcription: Ligand-dependent recruitment of estrogen receptor alpha to 2,3,7,8-tetrachlorodibenzo-p-dioxin-responsive promoters. Mol. Cell. Biol. 2005, 25, 5317–5328. [Google Scholar] [CrossRef] [PubMed]
- Ruegg, J.; Swedenborg, E.; Wahlstrom, D.; Escande, A.; Balaguer, P.; Pettersson, K.; Pongratz, I. The transcription factor aryl hydrocarbon receptor nuclear translocator functions as an estrogen receptor beta-selective coactivator, and its recruitment to alternative pathways mediates antiestrogenic effects of dioxin. Mol. Endocrinol. 2008, 22, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.C.; Cheng, L.C.; Lin, C.J.; Li, L.A. Dioxin and estrogen signaling in lung adenocarcinoma cells with different aryl hydrocarbon receptor/estrogen receptor alpha phenotypes. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Patrizi, B.; Siciliani de Cumis, M. TCDD Toxicity Mediated by Epigenetic Mechanisms. Int. J. Mol. Sci. 2018, 19, 4101. [Google Scholar] [CrossRef] [PubMed]
- Garrison, P.M.; Denison, M.S. Analysis of the murine AhR gene promoter. J. Biochem. Mol. Toxicol. 2000, 14, 1–10. [Google Scholar] [CrossRef]
- Garrison, P.M.; Rogers, J.M.; Brackney, W.R.; Denison, M.S. Effects of histone deacetylase inhibitors on the Ah receptor gene promoter. Arch. Biochem. Biophys. 2000, 374, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Beedanagari, S.R.; Taylor, R.T.; Bui, P.; Wang, F.; Nickerson, D.W.; Hankinson, O. Role of epigenetic mechanisms in differential regulation of the dioxin-inducible human CYP1A1 and CYP1B1 genes. Mol. Pharmacol. 2010, 78, 608–616. [Google Scholar] [CrossRef]
- Beedanagari, S.R.; Taylor, R.T.; Hankinson, O. Differential regulation of the dioxin-induced Cyp1a1 and Cyp1b1 genes in mouse hepatoma and fibroblast cell lines. Toxicol. Lett. 2010, 194, 26–33. [Google Scholar] [CrossRef]
- Ko, C.I.; Puga, A. Epigenetic mechanisms in AHR function. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 157–178. [Google Scholar]
- Kurita, H.; Schnekenburger, M.; Ovesen, J.L.; Xia, Y.; Puga, A. The Ah receptor recruits IKKalpha to its target binding motifs to phosphorylate serine-10 in histone H3 required for transcriptional activation. Toxicol. Sci. 2014, 139, 121–132. [Google Scholar] [CrossRef]
- Shen, E.S.; Whitlock, J.P., Jr. The potential role of DNA methylation in the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 1989, 264, 17754–17758. [Google Scholar] [PubMed]
- Wu, Q.; Ohsako, S.; Ishimura, R.; Suzuki, J.S.; Tohyama, C. Exposure of mouse preimplantation embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation status of imprinted genes H19 and Igf2. Biol. Reprod. 2004, 70, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Qiu, L.; Pu, Y.; Liu, C.; Zhang, X.; Wang, C.; Pu, W.; Fu, Y. Histone acetylation is involved in TCDDinduced cleft palate formation in fetal mice. Mol. Med. Rep. 2016, 14, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yuan, X.G.; Liu, C.P.; Zhai, S.N.; Zhang, D.W.; Fu, Y.X. Preliminary research on DNA methylation changes during murine palatogenesis induced by TCDD. J. Craniomaxillofac. Surg. 2017, 45, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.S.; Swanson, H.I. Dioxin-induced immortalization of normal human keratinocytes and silencing of p53 and p16INK4a. J. Biol. Chem. 2004, 279, 27187–27193. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yin, J.; Wu, W. Long non-coding RNA H19-mediated mouse cleft palate induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Exp. Ther. Med. 2016, 11, 2355–2360. [Google Scholar] [CrossRef] [PubMed]
- Moffat, I.D.; Boutros, P.C.; Celius, T.; Linden, J.; Pohjanvirta, R.; Okey, A.B. microRNAs in adult rodent liver are refractory to dioxin treatment. Toxicol. Sci. 2007, 99, 470–487. [Google Scholar] [CrossRef]
- Singh, N.P.; Singh, U.P.; Guan, H.; Nagarkatti, P.; Nagarkatti, M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS ONE 2012, 7, e45054. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H.; Alzahrani, A. MicroRNA-132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti-inflammation: A new Ahr-based exploration. Eur. J. Immunol. 2013, 43, 2771–2782. [Google Scholar]
- Mocarelli, P.; Gerthoux, P.M.; Ferrari, E.; Patterson, D.G., Jr.; Kieszak, S.M.; Brambilla, P.; Vincoli, N.; Signorini, S.; Tramacere, P.; Carreri, V.; et al. Paternal concentrations of dioxin and sex ratio of offspring. Lancet 2000, 355, 1858–1863. [Google Scholar] [CrossRef]
- Ryan, J.J.; Amirova, Z.; Carrier, G. Sex ratios of children of Russian pesticide producers exposed to dioxin. Environ. Health Perspect. 2002, 110, A699–A701. [Google Scholar] [CrossRef] [PubMed]
- ‘t Mannetje, A.; Eng, A.; Walls, C.; Dryson, E.; Kogevinas, M.; Brooks, C.; McLean, D.; Cheng, S.; Smith, A.H.; Pearce, N. Sex ratio of the offspring of New Zealand phenoxy herbicide producers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Occup. Environ. Med. 2017, 74, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Murray, F.J.; Smith, F.A.; Nitschke, K.D.; Humiston, C.G.; Kociba, R.J.; Schwetz, B.A. Three-generation reproduction study of rats given 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the diet. Toxicol. Appl. Pharmacol. 1979, 50, 241–252. [Google Scholar] [CrossRef]
- Ikeda, M.; Tamura, M.; Yamashita, J.; Suzuki, C.; Tomita, T. Repeated in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure affects male gonads in offspring, leading to sex ratio changes in F2 progeny. Toxicol. Appl. Pharmacol. 2005, 206, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Warita, K.; Tanida, T.; Sugawara, T.; Kitagawa, H.; Hoshi, N. Does paternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) affect the sex ratio of offspring? J. Vet. Med. Sci. 2007, 69, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Ohsako, S.; Tasaka, K.; Harayama, H.; Miyake, M.; Warita, K.; Tanida, T.; Mitsuhashi, T.; Nanmori, T.; Tabuchi, Y.; et al. When does the sex ratio of offspring of the paternal 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure decrease: In the spermatozoa stage or at fertilization? Reprod. Toxicol. 2010, 29, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; McConaha, M.; Boyd, K.L.; Osteen, K.G.; Bruner-Tran, K.L. Developmental dioxin exposure of either parent is associated with an increased risk of preterm birth in adult mice. Reprod. Toxicol. 2011, 31, 351–358. [Google Scholar] [CrossRef]
- You, Y.A.; Mohamed, E.A.; Rahman, M.S.; Kwon, W.S.; Song, W.H.; Ryu, B.Y.; Pang, M.G. 2,3,7,8-Tetrachlorodibenzo-p-dioxin can alter the sex ratio of embryos with decreased viability of Y spermatozoa in mice. Reprod. Toxicol. 2018, 77, 130–136. [Google Scholar] [CrossRef]
- Grech, V. Secular trends in newborn sex ratios. Early Hum. Dev. 2014, 90, 755–760. [Google Scholar] [CrossRef]
- Terrell, M.L.; Hartnett, K.P.; Marcus, M. Can environmental or occupational hazards alter the sex ratio at birth? A systematic review. Emerg. Health. Threats J. 2011, 4, 7109. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P.; Lehtiniemi, H.; Huusko, A.; Vahakangas, K.; Rautio, A. Polychlorinated biphenyls (PCBs) in relation to secondary sex ratio--a systematic review of published studies. Chemosphere 2013, 91, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.C.; Budinsky, R.A.; Aylward, L.L.; Faqi, A.S.; Carney, E.W. Sex ratio of the offspring of Sprague-Dawley rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in utero and lactationally in a three-generation study. Toxicol. Appl. Pharmacol. 2006, 216, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS ONE 2012, 7, e46249. [Google Scholar] [CrossRef] [PubMed]
- Manikkam, M.; Guerrero-Bosagna, C.; Tracey, R.; Haque, M.M.; Skinner, M.K. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS ONE 2012, 7, e31901. [Google Scholar] [CrossRef]
- Nilsson, E.; Larsen, G.; Manikkam, M.; Guerrero-Bosagna, C.; Savenkova, M.I.; Skinner, M.K. Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PLoS ONE 2012, 7, e36129. [Google Scholar] [CrossRef] [PubMed]
- Bruner-Tran, K.L.; Osteen, K.G. Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reprod. Toxicol. 2011, 31, 344–350. [Google Scholar] [CrossRef]
- Bruner-Tran, K.L.; Ding, T.; Yeoman, K.B.; Archibong, A.; Arosh, J.A.; Osteen, K.G. Developmental exposure of mice to dioxin promotes transgenerational testicular inflammation and an increased risk of preterm birth in unexposed mating partners. PLoS ONE 2014, 9, e105084. [Google Scholar] [CrossRef]
- Ding, T.; Mokshagundam, S.; Rinaudo, P.F.; Osteen, K.G.; Bruner-Tran, K.L. Paternal developmental toxicant exposure is associated with epigenetic modulation of sperm and placental Pgr and Igf2 in a mouse model. Biol. Reprod. 2018, 99, 864–876. [Google Scholar] [CrossRef]
- Sanabria, M.; Cucielo, M.S.; Guerra, M.T.; Dos Santos Borges, C.; Banzato, T.P.; Perobelli, J.E.; Leite, G.A.; Anselmo-Franci, J.A.; De Grava Kempinas, W. Sperm quality and fertility in rats after prenatal exposure to low doses of TCDD: A three-generation study. Reprod. Toxicol. 2016, 65, 29–38. [Google Scholar] [CrossRef]
- Olsvik, P.A.; Williams, T.D.; Tung, H.S.; Mirbahai, L.; Sanden, M.; Skjaerven, K.H.; Ellingsen, S. Impacts of TCDD and MeHg on DNA methylation in zebrafish (Danio rerio) across two generations. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2014, 165, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.R.; King-Heiden, T.C.; Peterson, R.E.; Heideman, W. Dioxin induction of transgenerational inheritance of disease in zebrafish. Mol. Cell. Endocrinol. 2014, 398, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.N.; Baker, B.B.; Baker, T.R. Ancestral TCDD Exposure Induces Multigenerational Histologic and Transcriptomic Alterations in Gonads of Male Zebrafish. Toxicol. Sci. 2018, 164, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, C.R. Minireview: Fetal-maternal hormonal signaling in pregnancy and labor. Mol. Endocrinol. 2009, 23, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Miller, D.C.; Harman, R.; Antczak, D.F.; Clark, A.G. Paternally expressed genes predominate in the placenta. Proc. Natl. Acad. Sci. USA 2013, 110, 10705–10710. [Google Scholar] [CrossRef]
- Bidgoli, S.A.; Karimi, M.; Asami, Z.; Baher, H.; Djamali Zavarhei, M. Association between testicular Aryl hydrocarbon Receptor levels and idiopathic male infertility: A case-control study in Iran. Sci. Total Environ. 2011, 409, 3267–3273. [Google Scholar] [CrossRef]
- Hansen, D.A.; Esakky, P.; Drury, A.; Lamb, L.; Moley, K.H. The aryl hydrocarbon receptor is important for proper seminiferous tubule architecture and sperm development in mice. Biol. Reprod. 2014, 90, 8. [Google Scholar] [CrossRef]
- Hurst, C.H.; DeVito, M.J.; Setzer, W.; Birnbaum, L.S. Acute administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in pregnant Long-Evans rats: Association of measured tissue concentrations with developmental effects. Toxicol. Sci. 2000, 53, 411–420. [Google Scholar] [CrossRef][Green Version]



{kind=link}
{kind=link}
{kind=link}
| A. Human Studies | ||||||
| Population | Dioxin Exposure | Effects Mediated via Paternal or Maternal Germline | Reference | |||
| Paternal Germline | Maternal Germline | Maternal and Paternal Germline | ||||
| Seveso population | Serum TCDD concentrations Unexposed: ≤15 pg/g fat Exposed: >15 pg/g fat Fathers: median 96.5, range 2.8–26,400 pg/g fat Mothers: median 62.8, range 6.45–12,500 pg/g fat | Male/female ratio ↓: Unexposed 55.7%, exposed 43.6% Fathers <19 years at exposure: Male/female ratio ↓: Unexposed 53.5%, exposed 38.2% | Male/female ratio not changed: 54.5% (ns) | Male/female ratio ↓: 44.2% | Mocarelli et al., 2000 [98] | |
| Russian pesticide producers | Serum TEQ concentration (mainly TCDD): Unexposed: not reported Exposed: median 243 pg/g fat, range 17–8520 pg/g fat | Male/female ratio ↓: Unexposed 51%, exposed 38%; higher exposed cohort with median 715 pg/g fat: 23% | Male/female ratio not changed (51%, ns) | Male/female ratio ↓: Unexposed 51%, exposed 40% | Ryan et al., 2002 [99] | |
| New-Zealand phenoxy herbicide producers | Serum TCDD concentration back-calculated to time of offspring’s birth (4 categories): <4, 4–20, 20–100 and ≥100 pg/g fat | Male/female ratio ↓: TCDD <20 pg/g fat: 60% TCDD ≥20 pg/g fat: 47% | Male/female ratio not changed: TCDD <20 pg/g fat: 53% TCDD ≥20 pg/g fat: 68%, ns | No data | Mannetje et al., 2017 [100] | |
| B. Experimental Studies | ||||||
| Species, Strain | Dosing of TCDD | Effects Mediated via Paternal or Maternal Germline | Reference | |||
| Dose | Timing | Paternal Germline | Maternal Germline | Maternal and Paternal Germline | ||
| Rat, Sprague Dawley | 0.1 µg/kg bw/day, in diet | 12 months starting 90 days prior to mating, TCDD exposed F0 males and females mated with unexposed partners | Cross-mating study: No harmful effects on pregnancy or resorptions. | Cross-mating study: No harmful effects on pregnancy, resorptions ↑ | Cross-mating study: Not examined | Murray et al., 1979 [101] |
| Rat, Holzman | Loading dose 400 ng/kg bw + maintenance doses 80 ng/kg bw/week Adipose tissue TCDD conc.: F0 dams GD 20: 1810, weaning: 840; F1 pups PND 28: ~480 pg/g wet weight | F0 females exposed 2 weeks before mating until end of lactation. TCDD exposed F1 males mated with unexposed females | F2: Male/female ratio ↓ (Ctr 52.2%, TCDD 38%). | Not examined | Not examined | Ikeda et al., 2005 [102] |
| Mouse, ICR | Loading dose 2 ng/kg bw + maintenance doses 5 × 0.4 ng/kg/bw/week or 2000 ng/kg bw + 5 × 400 ng/kg bw/week, oral gavage in sesame oil | 5 weeks before mating, TCDD exposed males mated with unexposed females | F1: Male/female ratio ↓: Ctr 53.1%, TCDD 2/0.4: 48.8% (ns), TCDD 2000/400: 46.2% | Not examined | Not examined | Ishihara et al., 2007 [103] |
| Mouse, ICR | Loading dose 2000 ng/kg bw + maintenance doses 5 × 400 ng/kg bw/week, oral gavage in sesame oil | 5 weeks before mating, TCDD exposed males mated with nonexposed females | Y-bearing/X-bearing sperm ratio ↓ (ns, Ctr: 2.68, TCDD: 2.36), sperm Sry DNA concentration ↓ (ns, Ctr 28.12, TCDD 25.80), male/female ratio of 2-cell embryos ↓ (Ctr: 53.95%, TCDD 47.92%) | Not examined | Not examined | Ishihara et al., 2010 [104] |
| Mouse, C57Bl/6 | 10 µg/kg bw, single dose, oral gavage in corn oil | GD 15.5 TCDD exposed F1 males mated with unexposed females and TCDD exposed F1 females mated with unexposed males | F: fertility ↓ (47% pregnant), premature births ↑ (Ctr 20 days, TCDD 18.5 days); placental weight ↓, pup weight ↓, placental progesterone receptor A and B ↓ and toll-like receptor-4 mRNA expression ↑, sensitivity to inflammation ↑ | F: fertility ↓ (39% pregnant); premature births ↑, pup weight ↓, placental progesterone receptor A and B ↓ and toll-like receptor-4 mRNA expression ↑, sensitivity to inflammation ↑ | F: fertility ↓ (0% pregnant) | Ding et al., 2011 [105] |
| Mouse, ICR | Epididymal sperm exposed to 0, 0.25, 25, or 2500 ng/mL in vitro | Incubation time 1 h | Sperm motility and viability concentration dependently ↓, acrosome-reacted spermatozoa ↑ at 25 and 2500 ng/mL, Y-spermatozoa survival concentration dependently ↓ at 25 and 2500 ng/mL, fertilization and early embryonic development in vitro ↓ at 25 and 2500 ng/mL, male/female ratio of 2-cell embryos dose-dependently ↓ at 0.25, 25, and 2500 ng/mL, male/female ratio of blastocysts concentration dependently ↓ at 25 and 2500 ng/mL | Not examined | Not examined | You et al., 2018 [106] |
| A. Rodent studies: both paternal and maternal exposure | ||||||
| Species, Strain | Exposure to TCDD | Effects | Reference | |||
| Dose | Timing | F1 Generation | F2 Generation | F3 Generation | ||
| Rat, Sprague Dawley | 0.001, 0.01 or 0.1 µg/kg bw/day, in diet | 90 days prior to mating throughout 3 generations (continuous exposure) | 0.001 µg/kg: slightly dilated renal pelvis ↑, 0.01 µg/kg: time from cohabitation to delivery ↑, fertility ↓, postnatal survival ↓, 0.1 µg/kg: fertility ↓, litter size ↓, gestation survival index ↓ (discontinued) | 0.001 µg/kg: no effects, 0.01 µg/kg: body weight ↓, time from cohabitation to delivery ↑, fertility ↓, litter size ↓, gestation survival index ↓, postnatal survival ↓ | 0.001 µg/kg: no effects, 0.01 µg/kg: body weight ↓, litter size ↓, gestation survival index ↓ | Murray et al., 1979 [101] |
| Rat, Sprague Dawley | 0.001, 0.01, or 0.1 µg/kg bw/day, in diet | 90 days prior to mating throughout 3 generations | Male/female ratio not changed | Male/female ratio not changed | Male/female ratio not changed | Rowlands et al., 2006 [110] (re-examination of the Murray et al. 1979 data 99]) |
| Rat, Sprague Dawley | 100 ng/kg/day ip in DMSO Total dose: 700 ng/kg/day | GD 8-14 | M: delayed puberty onset; testis weight ↑, prostate and kidney weight ↓ F: pubertal abnormalities; body weight ↓, ovarian primordial follicles ↓, polycystic ovary disease | Not examined | M: delayed puberty onset; kidney: weight ↓, fluid filled cysts, glomerular size ↓, thickening of Bowman’s capsule; serum testosterone ↑, 50 differentially methylated regions in sperm DNA, atrophic prostate duct epithelium F: early onset of puberty, kidney weight ↓, ovarian primordial follicles ↓, polycystic ovary disease | Manikkam et al., 2012a,b [111,112]; Nilsson et al., 2012 [113] |
| Rat, Sprague Dawley | 100 ng/kg/day ip in DMSO Total dose: 700 ng/kg/day | GD 8-14 | F: ovarian primordial follicles ↓, polycystic ovary disease: small ovarian cysts ↑ (ns), large ovarian cysts ↑ (ns) | Not examined | F: ovarian primordial follicles ↓, polycystic ovary disease: small ovarian cysts ↑, large ovarian cysts ↑ (ns) | Nilsson et al., 2012 [113] |
| B. Rodent studies: paternal or maternal exposure | ||||||
| Mouse, C57Bl/6 | 10 µg/kg, single dose, oral gavage in corn oil | GD 15.5 TCDD exposed F1 and F2 females mated with unexposed males | F: fertility ↓, premature births ↑, progesterone receptor immunostaining in uterus of infertile mice ↓, sensitivity to inflammation ↓ | F: fertility ↓, premature births ↑ | F: fertility ↓, premature births ↑ (in F4: progesterone receptor immunostaining in uterus of infertile mice ↓) | Bruner-Tran and Osteen, 2011 [114] |
| Mouse, C57Bl/6 | 10 µg/kg, single dose, oral gavage in corn oil | GD 15.5 TCDD exposed F1 and F2 males mated with unexposed females | M: fertility ↓ (47% pregnant), premature births in unexposed partners ↑, sperm concentration ↓, normal sperm morphology ↓, sperm AHR expression ↑, testicular inflammation and apoptosis ↑ | M: fertility ↓ (48% pregnant), premature births in unexposed partners ↑, normal sperm morphology ↓, sperm AHR expression ↑, testicular inflammation and apoptosis ↑ | M: fertility ↓ (50% pregnant), premature births in unexposed partners ↑, normal sperm morphology ↓, sperm AHR expression ↑, testicular inflammation and apoptosis ↑ | Bruner-Tran et al., 2014 [115] |
| Mouse, C57Bl/6 | 10 µg/kg, single dose, oral gavage in corn oil | GD 15.5 TCDD exposed F1 and F2 males mated with unexposed females | Placental weight ↓, pup weight ↓ F: 15% of genes in placenta differentially methylated, hypermethylation of progesterone receptor (Pgr), hypomethylation of insulin-like growth factor-2 (Igf2, ns), mRNA of Pgr-b, Pgr-a/b, Igf-2, and H19 ↓, IGR2 protein ↓, mRNA of DNA methyltransfereases ↑ Dnmt1, Dnmt3a (ns), Dnmt3b (ns) M: in sperm hypermethylation of Pgr, hypomethylation of Igf2 | Not examined | Placental weight ↓, pup weight ↓ F: 15% of genes in placenta differentially methylated, hypermethylation of Pgr, hypomethylation of Igf2 (ns). M: in sperm hypermethylation of Pgr (ns) and hypomethylation of Igf2 (ns), mRNA of Pgr-b, Pgr-a/b (ns), Igf-2 (ns) and H19 ↓, IGR2 protein ↓, mRNA of DNA methyltransfereases ↑ Dnmt1, Dnmt3a (ns), Dnmt3b (ns) | Ding et al., 2018 [116] |
| Rat, Wistar | 0.1, 0.5 or 1.0 µg/kg bw, single dose, oral gavage in corn oil | GD15 TCDD exposed F1 and F2 males mated with unexposed females | M: serum testosterone ↓ (dose-dependent, only 1.0 significant), sperm transit time ↓ (ns), normal sperm morphology ↓ (0.5 and 1.0) F: implants per corpora lutea ↓ in unexposed partners at 0.5 and 1.0 µg/kg bw: Ctr 75.2%, 0.5 62.0%, 1.0 58.7% | F: implants per corpora lutea ↓ in unexposed partners at 0.1, and 1.0 µg/kg bw: Ctr 61.9%, 0.1 41.1%, 0.5 50.5% (ns), 1.0 43.6% | F: implants per corpora lutea ↓ in unexposed partners at 0.1, 0.5 and 1.0 µg/kg bw: Ctr 82.4%, 0.1 50.7%, 0.5 56.6%, 1.0 31.8% | Sanabria et al., 2016 [117] |
| C. Zebrafish studies | ||||||
| F0 generation | F1 generation | F2 generation | ||||
| Zebrafish | 20 µg/kg in diet | Parental exposure 47 days | No effect on global DNA methylation in liver, CYP 1A1 ↑ | No effect on global DNA methylation in liver | Olsvik et al., 2014 [118] | |
| Zebrafish AB strain | 50 pg/mL in water (dissolved in DMSO) | 1 h at week 3 and week 7 post fertilization | Male/female ratio ↓ (Ctr 71.1%, TCDD 55.5%) F: atretic ovarian follicles (65.5%), egg release ↓, fertilization success ↓ Skeletal abnormalities (82.4%) axial kinks (54.5%) cranial malformations (46.9%) jaw malformations (34.5%) | Male/female ratio ↓ (Ctr 70.8%, TCDD 59.3%) F: atretic ovarian follicles (46.1%), egg release ↓ M: elicitation of egg release ↓, fertilization success ↓ Skeletal abnormalities (34.9%) axial kinks (28.1%) cranial malformations (11.7%) jaw malformations (3.7%, ns) | Male/female ratio ↓ (Ctr 78.7%, TCDD 61.4%) F: atretic ovarian follicles (7.7%, ns) M: elicitation of egg release ↓, fertilization success ↓ Skeletal abnormalities (22.1%) axial kinks (17.3%) cranial malformations (7.8%, ns) jaw malformations (0.9%, ns) | Baker et al., 2014 [119] |
| Zebrafish AB strain | 50 pg/mL in water (dissolved in DMSO) | 1 h at week 3 and week 7 post fertilization | M: in testis 722 differentially expressed genes | M: in seminiferous tubules spermatogonia ↑, spermatozoa ↓, in testis 634 differentially expressed genes | M: in seminiferous tubules spermatozoa ↓ (ns), in testis 1105 differentially expressed genes | Meyer et al., 2018 [120] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viluksela, M.; Pohjanvirta, R. Multigenerational and Transgenerational Effects of Dioxins. Int. J. Mol. Sci. 2019, 20, 2947. https://doi.org/10.3390/ijms20122947
Viluksela M, Pohjanvirta R. Multigenerational and Transgenerational Effects of Dioxins. International Journal of Molecular Sciences. 2019; 20(12):2947. https://doi.org/10.3390/ijms20122947
Chicago/Turabian StyleViluksela, Matti, and Raimo Pohjanvirta. 2019. "Multigenerational and Transgenerational Effects of Dioxins" International Journal of Molecular Sciences 20, no. 12: 2947. https://doi.org/10.3390/ijms20122947
APA StyleViluksela, M., & Pohjanvirta, R. (2019). Multigenerational and Transgenerational Effects of Dioxins. International Journal of Molecular Sciences, 20(12), 2947. https://doi.org/10.3390/ijms20122947