The Role of Nitric Oxide, ADMA, and Homocysteine in The Etiopathogenesis of Preeclampsia—Review



Abstract
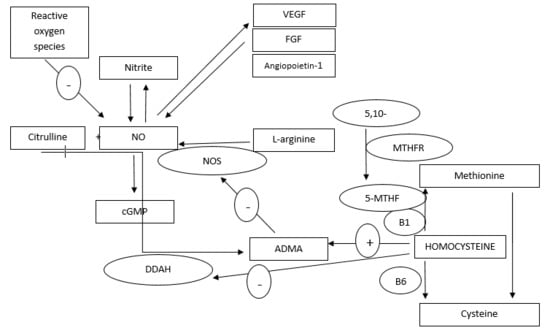
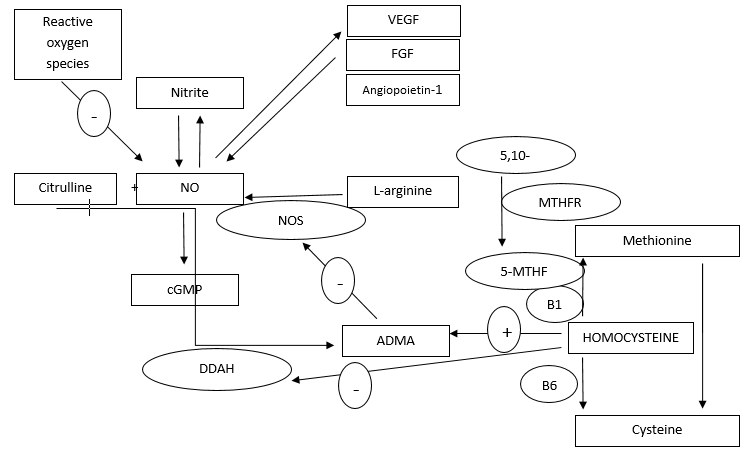{kind=link}
{kind=link}
1. Preeclampsia—Background
2. Metabolism and Biological Role of NO, ADMA, and Homocysteine
3. NO, ADMA and Homocysteine in Pregnancy
3.1. ADMA in Pregnancy
3.2. Homocysteine and Pregnancy
4. NO, ADMA, and Homocysteine in Preeclampsia
4.1. NO Pathway Dysfunction in PE
4.2. Homocysteine in PE
5. Therapeutic Potential of NO Pathway during Pregnancy
6. Conclusions
Funding
Conflicts of Interest
References
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens 2014, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy. Hypertension in Pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Tranquilli, A.L.; Brown, M.A.; Zeeman, G.G.; Dekker, G.; Sibai, B.M. The definition of severe and early-onset preeclampsia. Statements from the International Society for the Study of Hypertension in Pregnancy (ISSHP). Pregnancy Hypertens. 2013, 3, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Taylor, R.N.; Musci, T.J.; Rodgers, G.M.; Hubel, C.A.; McLaughlin, M.K. Preeclampsia: An endothelial cell disorder. Am. J. Obstet. Gynecol. 1989, 161, 1200–1204. [Google Scholar] [CrossRef]
- Roberts, J.M.; Hubel, C.A. The two stage model of preeclampsia: Variations on the theme. Placenta 2009, 30, S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.C.; Werb, Z.; Fisher, S.K. Implantation and the placenta: Key pieces of the development puzzle. Science 1994, 266, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.A.; Robertson, W.B.; Dixon, H.G. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet. Gynecol. Annu. 1972, 1, 177–191. [Google Scholar] [CrossRef]
- Huppertz, B. Placental origins of preeclampsia: Challenging the current hypothesis. Hypertension 2008, 51, 970–975. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt-1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Romero, R.; Kim, Y.M.; Kim, G.J.; Kim, M.R.; Espinoza, J.; Bujold, E.; Goncalves, L.; Gomez, R.; Edwin, S.; et al. Plasma soluble vascular endothelial growth factor receptor -1 concentration is elevated prior to the cinical diagnosis of preeclampsia. J. Matern. Fetal Neonatal Med. 2005, 17, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Dymara-Konopka, W.; Laskowska, M.; Blazewicz, A. Angiogenic Imbalance as a Contributor of Preeclampsia. Curr. Pharm. Biotechnol. 2018, 19, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Harihana, N.; Shoemker, A.; Wagner, S. Pathophysiology of hypertension in preeclampsia. Clin. Pract. 2016, 13, 33–37. [Google Scholar]
- Karumanchi, S.A.; Maynard, S.E.; Stillman, I.E.; Epstein, F.H.; Sukhatme, V.P. Preeclampsia: A renal perspective. Kidney Int. 2005, 67, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Escudero, C. The Placenta in Preeclampsia. Pregnancy Hypertens 2012, 2, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Ferrazzi, E.; Zullino, S.; Stampalija, T.; Vener, C.; Cavoretto, P.; Gervasi, M.T.; Vergani, P.; Mecacci, F.; Marozio, L.; Oggè, G.; et al. Bedside diagnosis of two major clinical phenotypes of hypertensive disorders of pregnancy. Ultrasound Obstet. Gynecol. 2016, 48, 224–231. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L.; Staff, A.C. IFPA Senior Award Lecture: Making sense of pre-eclampsia. Placenta 2014, 35, S20–S25. [Google Scholar] [CrossRef]
- Huang, L.T.; Hsieh, C.S.; Chang, K.A.; Tain, Y.L. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int. J. Mol. Sci. 2012, 13, 14606–14622. [Google Scholar] [CrossRef]
- Baylis, C.; Beinder, E.; Suto, T.; August, P. Recent insights into the roles of nitric oxide and renin-angiotensin in the pathophysiology of preeclamptic pregnancy. Semin. Nephrol. 1998, 18, 208–230. [Google Scholar]
- Lowe, D.T. Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric Oxide 2000, 4, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Demir, B.; Demir, S.; Pasa, S. The role of homocysteine, asymmetric dimethylarginine and nitric oxide in preeclampsia. J. Obstet. Gynaecol. 2012, 32, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Khalil, R.A.; Granger, J.P. Vascular mechanisms of increased arterial pressure in preeclampsia: Lessons from animal models. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R29–R45. [Google Scholar] [CrossRef] [PubMed]
- Fickling, S.A.; Williams, D.; Vallance, P.; Nussey, S.S.; Whitley, G.S. Plasma concentrations of endogenous inhibitor of nitric oxide synthesis in normal pregnancy and preeclampsia. Lancet 1993, 342, 242–243. [Google Scholar] [CrossRef]
- Speer, P.D.; Powers, R.W.; Frank, M.P.; Harger, G.; Markovic, N.; Roberts, J.M. Elevated asymmetric dimethylarginine concentrations precede clinical preeclampsia, but not pregnancies with small-for-gestational-age infants. Am. J. Obstet. Gynecol. 2008, 198, 112 e111–112 e117. [Google Scholar] [CrossRef]
- Holden, D.P.; Fickling, S.A.; Whitley, G.S.; Nussey, S.S. Plasma concentrations of asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and preeclampsia. Am. J. Obstet. Gynecol. 1998, 178, 551–556. [Google Scholar] [CrossRef]
- Pettersson, A.; Hedner, T.; Milsom, I. Increased circulating concentrations of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthesis, in preeclampsia. Acta Obstet. Gynecol. Scand. 1998, 77, 808–813. [Google Scholar] [CrossRef]
- Aubard, Y.; Darodes, N.; Cantaloube, M. Hyperhomocysteinemia and pregnancy—Review of our present understanding and therapeutic implications. Eur. J. Obstet. Gynecol. Reprod. Biol. 2000, 93, 157–165. [Google Scholar] [CrossRef]
- Stühlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef]
- Herrmann, W.; Isber, S.; Obeid, R.; Herrmann, M.; Jouma, M. Concentrations of homocysteine, related metabolites and asymmetric dimethylarginine in preeclamptic women with poor nutritional status. Clin. Chem. Lab. Med. 2005, 43, 1139–1146. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzk, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Nitric oxide. A novel signal transduction mechanism for transcellular communication. Hypertension 1990, 16, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug. Discovery 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, J.; Pastore, M.B.; Magness, R.R. Endothelial caveolar subcellular domain regulation of endothelial nitric oxide synthase. Clin. Exp. Pharmacol. Physiol. 2013, 40, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Gielen, S.; Sandri, M.; Erbs, S.; Adams, V. Exercise-induced modulation of endothelial nitric oxide production. Curr. Pharm. Biotechnol. 2011, 12, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Quillon, A.; Fromy, B.; Debret, R. Endothelium microenvironment sensing leading to nitric oxide mediated vasodilation: A review of nervous and biomechanical signals. Nitric Oxide 2015, 45, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Sladek, S.M.; Magness, R.R.; Conrad, K.P. Nitric oxide and pregnancy. Am. J. Physiol. 1997, 272, R441–R463. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Mori, C.; Yoshikawa, H.; Miyazaki, Y.; Kansaku, N.; Tanaka, K.; Morita, H.; Takizawa, T. Changes in nitric oxide production levels and expression of nitric oxide synthase isoforms in the rat uterus during pregnancy. Biosci. Biotechnol. Biochem. 2009, 73, 2163–2166. [Google Scholar] [CrossRef]
- Purcell, T.L.; Given, R.; Chwalisz, K.; Garfield, R.E. Nitric oxide synthase distribution during implantation in the mouse. Mol. Hum. Reprod. 1999, 5, 467–475. [Google Scholar] [CrossRef]
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol. 2006, 572, 25–30. [Google Scholar] [CrossRef]
- Shizukuda, Y.; Tang, S.; Yokota, R.; Ware, J.A. Vascular endothelial growth factor-induced endothelial cell migration and proliferation depend on a nitric oxide-mediated decrease in protein kinase C activity. Circ. Res. 1999, 85, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Stallmeyer, B.; Kampfer, H.; Schaffner, C.; Pfeilschifter, J. Differential regulation of vascular endothelial growth factor and its receptor fms-like-tyrosine kinase is mediated by nitric oxide in rat renal mesangial cells. Biochem. J. 1999, 338, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kulandavelu, S.; Whiteley, K.J.; Qu, D.; Mu, J.; Bainbridge, S.A.; Adamson, S.L. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 2012, 60, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.; Jozkowicz, J. Regulation of vascular endothelial growth factor synthesis by nitric oxide: Facts and controversies. Antioxid. Redox. Signal. 2003, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef]
- Brustolin, S.; Giugliani, R.; Félix, T.M. Genetics of homocysteine metabolism and associated disorders. Braz. J. Med. Biol. Res. 2010, 43, 1–7. [Google Scholar] [CrossRef]
- Walker, M.; Smith, G.; Perkins, S.; Keely, E.; Garner, P. Changes in homocysteine levels during normal pregnancy. Am. J. Obstet. Gynecol. 1999, 180, 660–664. [Google Scholar] [CrossRef]
- Bailey, L.; Gregory, J. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: Metabolic significance, risks and impact on folate requirement. J. Nutr. 1999, 129, 919–922. [Google Scholar] [CrossRef]
- Goyette, P.; Sumner, J.; Milos, R.; Duncan, A.; Rosenblatt, D.; Matthews, R.; Rozen, R. Human methylenetetrahydrofolate reductase: Isolation of cDNA, mapping, and mutation identification. Nat. Genet. 1994, 7, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Faeh, D.; Chiolero, A.; Paccaud, F. Homocysteine as a risk factor for cardiovascular disease: Should we (still) worry about it? Swiss Med. Wkly. 2006, 136, 745–756. [Google Scholar] [PubMed]
- Baszczuk, A.; Kopczynski, Z. Hyperhomocysteinemia in patients with cardiovascular disease. Postepy Hig. Med. Dosw. 2014, 68, 579. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Eikelboom, J.W. Homocysteine and vascular disease. Lancet 1999, 354, 407–413. [Google Scholar] [CrossRef]
- Clarke, R.; Woodhouse, P.; Ulvik, A.; Frost, C.; Sherliker, P.; Refsum, H. Variability and determinants of total homocysteine concentrations in plasma in an elderly population. Clin. Chem. 2010, 44, 102–107. [Google Scholar]
- Thrombosis Interest Group of Canada. Thrombophilia: Homocysteinemia and Methylene Tetrahydrofolate Reductase. Available online: http://thrombosiscanada.ca/?page_id=18# (accessed on 17 June 2015).
- Curro, M.; Gugliandolo, A.; Gangemi, C.; Risitano, R.; Ientile, R.; Caccamo, D. Toxic effects of mildly elevated homocysteine concentrations in neuronal-like cells. Neurochem. Res. 2014, 39, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, I.S.; Jacques, P.F.; Selhub, J.; Bostom, A.G.; Chen, Z.; Curtis Ellison, R.; Eckfeldt, J.H.; Rozen, R. The 1298A→C polymorphism in methylenetetrahydrofolate reductase (MTHFR): In vitro expression and association with homocysteine. Atherosclerosis 2001, 156, 409–415. [Google Scholar] [CrossRef]
- Stanger, O.; Herrmann, W.; Pietrzik, K.; Fowler, B.; Geisel, J.; Dierkes, J.; Weger, M. Clinical use and rational management of homocysteine, folic acid, and B vitamins in cardiovascular and thrombotic diseases. Z. Kardiol. 2004, 93, 439–453. [Google Scholar] [CrossRef]
- Ntaios, G.; Savopoulos, C.; Grekas, P.; Hatzitolios, A. The controversial role of B-vitamins in cardiovascular risk: An update. Arch. Cardiovasc. Dis. 2009, 102, 847–854. [Google Scholar] [CrossRef]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111. [Google Scholar]
- Boushey, C.J.; Beresford, S.A.; Omenn, G.S.; Motulsky, A.G. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995, 274, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Ueland, P.M.; Nygard, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef] [PubMed]
- Guilland, J.; Favier, A.; Potier de Courcy, G.; Galan, P.; Hercberg, S. Hyperhomocysteinemia: An independent risk factor or a simple marker of vascular disease?. 1. Basic data. Pathol. Biol. 2003, 51, 101–110. [Google Scholar] [CrossRef]
- Wong, Y.Y.; Golledge, J.; Flicker, L.; McCaul, K.A.; Hankey, G.J.; van Bockxmeer, F.M.; Yeap, B.B.; Norman, P.E. Plasma total homocysteine is associated with abdominal aortic aneurysm and aortic diameter in older men. J. Vasc. Surg. 2013, 58, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, X.; Kong, W. Hyperhomocysteinaemia and vascular injury: Advances in mechanisms and drug targets. Br. J. Pharmacol. 2017, 175, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.F.; Wolf, P.A. Plasma Homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef]
- McIlroy, S.P.; Dynan, K.B.; Lawson, J.T.; Patterson, C.C.; Passmore, A.P. Moderately elevated plasma homocysteine, methylenetetrahydrofolate reductase genotype, and risk for stroke, vascular dementia, and Alzheimer disease in Northern Ireland. Stroke 2002, 33, 2351–2356. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Clausen, C.; Córdoba-Porras, A.; Bedoya, G.; Silva, F.; Zarruk, J.; López-Jaramillo, P.; Villa, L. Increased plasma levels of total homocysteine but not asymmetric dimethylarginine in Hispanic subjects with ischemic stroke FREC-VI sub-study. Eur. J. Neurol. 2012, 19, 417–425. [Google Scholar] [CrossRef]
- Herrmann, M.; Widmann, T.; Herrmann, W. Homocysteine–a newly recognised risk factor for osteoporosis. Clin. Chem. Lab. Med. 2005, 43, 1111–1117. [Google Scholar] [CrossRef]
- Perna, A.F.; Sepe, I.; Lanza, D.; Pollastro, R.M.; De Santo, N.G.; Ingrosso, D. Hyperhomocysteinemia in chronic renal failure: Alternative therapeutic strategies. J. Ren. Nutr. 2012, 22, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Brattström, L.; Wilcken, D.E. Homocysteine and cardiovascular disease: Cause or effect? Am. J. Clin. Nutr. 2000, 72, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.H.; Steinsland, O.S.; Wang, Y.; Yallampalli, C.; Dong, Y.L.; Sanchez, J.M. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ. Res. 2000, 87, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Kream, R.M. Reciprocal regulation of cellular nitric oxide formation by nitric oxide synthase and nitrite reductases. Med. Sci. Monit. 2011, 17, RA221–RA226. [Google Scholar] [CrossRef] [PubMed]
- Sanghavi, M.; Rutherford, J.D. Cardiovascular physiology of pregnancy. Circulation 2014, 130, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Leiva, A.; Fuenzalida, B.; Barros, E.; Sobrevia, B.; Salsoso, R.; Sáez, T.; Villalobos, R.; Silva, L.; Chiarello, I.; Toledo, F.; et al. Nitric oxide is a central common metabolite in vascular dysfunction associated with diseases of human pregnancy. Curr. Vasc. Pharmacol. 2016, 14, 237–259. [Google Scholar] [CrossRef]
- Conrad, K.P.; Kerchner, L.J.; Mosher, M.D. Plasma and 24-h NO(x) and cGMP during normal pregnancy and preeclampsia in women on a reduced NO(x) diet. Am. J. Physiol. 1999, 277, F48–F57. [Google Scholar]
- Buhimschi, I.; et al. Involvement of a nitric oxide-cyclic guanosine monophosphate pathway in control of human uterine contractility during pregnancy. Am. J. Obstet. Gynecol. 1995, 172, 1577–1584. [Google Scholar] [CrossRef]
- Baylis, C.; Vallance, P. Measurement of nitrite and nitrate levels in plasma and urine: What does this measure tell us about the activity of the endogenous nitric oxide system? Curr. Opin. Nephrol. Hypertens. 1998, 7, 59–62. [Google Scholar] [CrossRef]
- Choi, J.W.; Im, M.W.; Pai, S.H. Nitric oxide production increases during normal pregnancy and decreases in preeclampsia. Ann. Clin. Lab. Sci. 2002, 32, 257–263. [Google Scholar]
- Jo, T.; Takauchi, Y.; Nakajima, Y.; Fukami, K.; Kosaka, H.; Terada, N. Maternal or umbilical venous levels of nitrite/nitrate during pregnancy and at delivery. In Vivo 1998, 12, 523–526. [Google Scholar] [PubMed]
- Shaamash, A.H.; Elsnosy, E.D.; Makhlouf, A.M.; Zakhari, M.M.; Ibrahim, O.A.; EL-dien, H.M. Maternal and fetal serum nitric oxide (NO) concentrations in normal pregnancy, pre-eclampsia and eclampsia. Int. J. Gynaecol. Obstet. 2000, 68, 207–214. [Google Scholar] [CrossRef]
- Hata, T.; Hashimoto, M.; Kanenishi, K.; Akiyama, M.; Yanagihara, T.; Masumura, S. Maternal circulation nitrite levels are decreased in both normal normotensive pregnancies and pregnancies with preeclampsia. Gynecol. Obstet. Invest. 1999, 48, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Tibben, E.; Zammit, V.C.; Cario, G.M.; Carlton, M.A. Nitric oxide excretion in normal and hypertensive pregnancies. Hyperten. Pregnancy 1995, 14, 319–326. [Google Scholar] [CrossRef]
- Smarason, A.K.; Allman, K.G.; Young, D.; Redman, C.W.G. Elevated levels of serum nitrate, a stable end product of nitric oxide, in women with preeclampsia. Br. J. Obstet. Gynecol. 1997, 104, 538–543. [Google Scholar] [CrossRef]
- Shah, D.A.; Khalil, R.A. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem. Pharmacol. 2015, 95, 211–226. [Google Scholar] [CrossRef]
- Vida, G.; Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Böger, S.M. Birth by cesarean section is associated with elevated neonatal plasma levels of dimethylarginines. Pediatr. Int. 2012, 54, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Hultberg, B.; Brattström, L.; Isaksson, A. Decreased serum homocysteine in pregnancy. Eur. J. Clin. Chem. Clin. Biochem. 1992, 30, 377–379. [Google Scholar]
- Hague, B.; Whiting, M.; Tallis, G. South Australian experience with hyperhomocysteinaemia as a risk factor in obstetrics. Time for a trial? Neth. J. Med. 1997, 52, S24. [Google Scholar]
- Malinow, M.R.; Rajkovic, A.; Duell, P.B.; Hess, D.L.; Upson, B.M. The relationship between maternal and neonatal umbilical cord plasma homocysteine suggests a potential role for maternal homocysteine in fetal metabolism. Obstet. Gynecol. 1998, 178, 228–233. [Google Scholar]
- Steegers-Theunissen, R.; Wathen, N.; Eskes, T.; Van Raaij-Selten, B.; Chard, T. Maternal and fetal levels of methionine and homocysteine in early human pregnancy. Br. J. Obstet. Gynecol. 1997, 104, 20–24. [Google Scholar] [CrossRef]
- Lopez-Quesada, E.; Vilaseca, M.A.; Lailla, J.M. Plasma total homocysteine in uncomplicated pregnancy and in preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 108, 45–49. [Google Scholar] [CrossRef]
- Bergen, N.E.; Jaddoe, V.W.; Timmermans, S.; Hofman, A.; Lindemans, J.; Russcher, H.; Raat, H.; SteegersTheunissen, R.P.; Steegers, E.A. Homocysteine and folate concentrations in early pregnancy and the risk of adverse pregnancy outcomes: The Generation R Study. BJOG 2012, 119, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.L.; McPartlin, J.M.; Kirke, P.N.; Lee, Y.J.; Conley, M.R.; Weir, D.G.; Scott, J.M. Homocysteine metabolism in pregnancies complicated by neuraltube defects. Lancet 1995, 345, 149–151. [Google Scholar] [CrossRef]
- Chen, H.; Yang, X.; Lu, M. Methylenetetrahydrofolate reductase gene polymorphisms and recurrent pregnancy loss in China: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2016, 293, 283–290. [Google Scholar] [CrossRef] [PubMed]
- De Falco, M.; Pollio, F.; Scaramelino, M.; Pontillo, M.; Lieto, A.D. Homocysteinemia during pregnancy and placental disease. Clin. Exp. Obstet. Gynecol. 2000, 27, 188–190. [Google Scholar] [PubMed]
- Steegers-Theunissen, R.P.; Van Iersel, C.A.; Peer, P.G.; Nelen, W.L.; Steegers, E.A. Hyperhomocysteinemia, pregnancy complications, and the timing of investigation. Obstet. Gynecol. 2004, 104, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Seligman, S.P.; et al. The role of nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 944–948. [Google Scholar] [CrossRef]
- Mutlu-Turkoglu, U.; Aykac-Toker, G.; Ibrahimoglu, L.; Ademoglu, E.; Uysal, M. Plasma nitric oxide metabolites and lipid peroxide levels in preeclamptic pregnant women before and after delivery. Gynecol. Obstet. Invest. 1999, 48, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.K.; et al. Evaluation of nitric oxide as a mediator of severe preeclampsia. Am. J. Obstet. Gynecol. 1996, 175, 1013–1017. [Google Scholar] [CrossRef]
- Schiessl, B.; Strasburger, C.; Bidlingmaier, M.; Mylonas, I.; Jeschke, U.; Kainer, F.; Friese, K. Plasma- and urine concentrations of nitrite/nitrate and cyclic Guanosinemono phosphate in intrauterine growth restricted and preeclamptic pregnancies. Arch. Gynecol. Obstet. 2006, 274, 150–154. [Google Scholar] [CrossRef]
- Pathak, N.; et al. Estimation of oxidative products of nitric oxide (nitrates, nitrites) in preeclampsia. Aust. N. Z. J. Obstet. Gynaecol. 1999, 39, 484–487. [Google Scholar] [PubMed]
- Alexander, B.T.; Miller, M.T.; Kassab, S.; Novak, J.; Reckelhoff, J.F.; Kruckeberg, W.C.; Granger, J.P. Differential expression of renal nitric oxide synthase isoforms during pregnancy in rats. Hypertension 1999, 33, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.K.; English, F.A.; Johns, E.J.; Kenny, L.C. Plasma-Mediated Vascular Dysfunction in the Reduced Uterine Perfusion Pressure Model of Preeclampsia: A Microvascular Characterization. Hypertension 2009, 54, 345–351. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Dejam, A.; Lauer, T.; Rassaf, T.; Schindler, A.; Picker, O.; Scheeren, T.; Godecke, A.; Schrader, J.; Schulz, R.; et al. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic. Biol. Med. 2003, 35, 790–796. [Google Scholar] [CrossRef]
- Kelm, M.; Preik-Steinhoff, H.; Preik, M.; Strauer, B.E. Serum nitrite sensitively reflects endothelial NO formation in human forearm vasculature: Evidence for biochemical assessment of the endothelial L-arginine-NO pathway. Cardiovasc. Res. 1999, 41, 765–772. [Google Scholar] [CrossRef]
- Pimentel, A.M.; Pereira, N.R.; Costa, C.A.; Mann, G.E.; Cordeiro, V.S.; de Moura, R.S.; Brunini, T.M.C.; Mendes-Ribeiro, A.C.; Resende, Â.C. L-arginine-nitric oxide pathway and oxidative stress in plasma and platelets of patients with pre-eclampsia. Hypertens. Res. 2013, 36, 783–788. [Google Scholar] [CrossRef]
- Eleuterio, N.M.; Palei, A.C.; Machado, J.S.R.; Tanus-Santos, J.E.; Cavalli, R.C.; Sandrim, V.C. Relationship between adiponectin and nitrite in healthy and preeclampsia pregnancies. Clin. Chim. Acta. 2013, 423, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Sandrim, V.C.; Palei, A.C.; Metzger, I.F.; Gomes, V.A.; Cavalli, R.C.; Tanus-Santos, J.E. Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension 2008, 52, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Groesch, K.A.; Torry, R.J.; Wilber, A.C.; Abrams, R.; Bieniarz, A.; Guilbert, L.J.; Torry, D.S. Nitric oxide generation affects pro- and anti-angiogenic growth factor expression in primary human trophoblast. Placenta 2011, 32, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, M.; Chen, Y.; Wang, S. Homocysteine, endothelin-1 and nitric oxide in patients with hypertensive disorders complicating pregnancy. Int. J. Clin. Exp. Pathol. 2015, 8, 15275–15279. [Google Scholar]
- Cadnapaphornchai, M.A.; Ohara, M.; Morris, K.G.; Knotek, M.; Rogachev, B.; Ladtkow, T.; Carter, E.P.; Schrier, R.W. Chronic NOS inhibition reverses systemic vasodi- lation and glomerular hyperfiltration in pregnancy. Am. J. Physiol. Ren. Physiol. 2001, 280, 592–598. [Google Scholar] [CrossRef]
- Laskowska, M.; Laskowska, K.; Oleszczuk, J. The relation of maternalserum eNOS, NOSTRIN and ADMA levels with aetiopathogenesis of preeclampsia and/or intrauterine fetal growth restriction. J. Matern. Fetal. Neonatal. Med. 2015, 28, 26–32. [Google Scholar] [CrossRef]
- Laskowska, M.; Laskowska, K.; Oleszczuk, J. PP135. Maternal serum levels of endothelial nitric oxide synthase and ADMA, an endogenous ENOS inhibitor in pregnancies complicated by severe preeclampsia. Pregnancy Hypertens. 2012, 2, 312. [Google Scholar] [CrossRef]
- Myatt, L.; Eis, A.L.; Brockman, D.E.; Greer, I.A.; Lyall, F. Endothelial nitric oxide synthase in placental villous tissue from normal, pre-eclamptic and intrauterine growth restricted pregnancies. Hum. Reprod. 1997, 12, 167–172. [Google Scholar] [CrossRef]
- Smith-Jackson, K.; Hentschke, M.R.; Poli-de-Figueiredo, C.E.; da Costa, B.P.; Kurlak, L.O.; Pipkin, F.B.; Czajkad, A.; Mistry, H. D. Placental expression of eNOS, iNOS and the major protein components of caveolae in women with preeclampsia. Placenta 2015, 36, 607–610. [Google Scholar] [CrossRef]
- Beinder, E.; Mohaupt, M.G.; Schlembach, D.; Fischer, T.; Sterzel, R.B.; Lang, N.; Baylis, C. Nitric oxide synthase activity and Doppler parameters in the fetoplacental and uteroplacental circulation in preeclampsia. Hypertens. Pregnancy 1999, 18, 115–127. [Google Scholar] [CrossRef]
- Orange, S.J.; et al. Placental endothelial nitric oxide synthase localization and expression in normal human pregnancy and preeclampsia. Clin. Exp. Pharmacol. Physiol. 2003, 30, 376–381. [Google Scholar] [CrossRef]
- Marshall, S.A.; Hannan, N.J.; Jelinic, M.; Nguyen, T.P.; Girling, J.E.; Parry, L.J. Animal models of preeclampsia: Translational failings and why. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R499–R508. [Google Scholar] [CrossRef]
- Gonçalves-Rizzi, V.H.; Possomato-Vieira, J.S.; Graça, T.U.S.; Nascimento, R.A.; Dias-Junior, C.A. Sodium nitrite attenuates hypertension- in-pregnancy and blunts increases in soluble fms-like tyrosine kinase-1 and in vascular endothelial growth factor. Nitric Oxide 2016, 57, 71–78. [Google Scholar]
- Khalil, A.A.; Tsikas, D.; Akolekar, R.; Jordan, J.; Nicolaides, K.H. Asymmetric dimethylarginine, arginine and homoarginine at 11-13 weeks’ gestation and preeclampsia: A case-control study. J. Hum. Hypertens. 2013, 27, 38–43. [Google Scholar] [CrossRef] [PubMed]
- López‐Alarcón, M.; Montalvo‐Velarde, I.; Vital‐Reyes, V.S.; Hinojosa‐Cruz, J.C.; Leaños‐Miranda, A.; Martínez‐Basila, A. Serial determinations of asymmetric dimethylarginine and homocysteine during pregnancy to predict pre-eclampsia: A longitudinal study. BJOG 2015, 122, 1586–1592. [Google Scholar]
- Zheng, J.J.; Wang, H.O.; Huang, M.; Zheng, F.Y. Assessment of ADMA, estradiol, and progesterone in severe preeclampsia. Clin. Exp. Hypertens. 2016, 38, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Laskowska, M.; Laskowska, K.; Terbosh, M.; Oleszczuk, J. A comparison of maternal serum levels of endothelial nitric oxide synthase, asymmetric dimethylarginine, and homocysteine in normal and preeclamptic pregnancies. Med. Sci. Monit. 2013, 19, 430–437. [Google Scholar]
- Alpoim, P.N.; Godoi, L.C.; Freitas, L.G.; Gomes, K.B.; Dusse, L.M. Assessment of L-arginine asymmetric 1 dimethyl (ADMA) in early-onset and late-onset (severe) preeclampsia. Nitric Oxide 2013, 33, 81–82. [Google Scholar] [CrossRef]
- Bian, Z.; Shixia, C.; Duan, T. First-trimester maternal serum levels of sFLT1, PGF and ADMA predict preeclampsia. PLoS One 2015, 10, e0124684. [Google Scholar] [CrossRef]
- Rizos, D.; Eleftheriades, M.; Batakis, E.; Rizou, M.; Halisassos, A.; Hassiakos, D.; Botsis, D. Levels of asymmetric dimethylarginine throughout normal pregnancy and in pregnancies complicated by preeclampsia or had a small for gestational age baby. J. Matern. Fetal. Neonatal. Med. 2012, 25, 1311–1315. [Google Scholar] [CrossRef]
- Savvidou, M.D.; Hingorani, A.D.; Tsikas, D.; Frolich, J.C.; Vallance, P.; Nicolaides, K.H. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet 2003, 361, 1511–1517. [Google Scholar] [CrossRef]
- Kim, M.W.; Hong, S.C.; Choi, J.S.; Han, J.Y.; Oh, M.J.; Kim, H.J.; Nava-Ocampo, A.; Koren, G. Homocysteine, folate and pregnancy outcomes. J. Obstet. Gynaecol. 2012, 32, 520–524. [Google Scholar] [CrossRef]
- Dekker, A.G.; DeVries, J.I.P.; Doelitzsch, P.M.; Huijgens, P.C.; von Blomberg, B.M.; Jakobs, C.; van Geijn, H.P. Underlying disorders associated with severe early onset preeclampsia. Am. J. Obstet. Gynecol. 1995, 173, 1042–1048. [Google Scholar] [CrossRef]
- Acilmis, Y.G.; Dikensoy, E.; Kutlar, A.I.; Balat, O.; Cebesoy, F.B.; Ozturk, E.; Cicek, H.; Pence, S. Homocysteine, folic acid and vitamin B12 levels in maternal and umbilical cord plasma and homocysteine levels in placenta in pregnant women with preeclampsia. J. Obstet. Gynaecol. Res. 2011, 37, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Mujawar, S.A.; Patil, V.W.; Daver, R.G. Study of serum homocysteine, folic Acid and vitamin B(12) in patients with preeclampsia. Indian J. Clin. Biochem. 2011, 26, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Guven, M.A.; Coskun, A.; Ertas, I.E.; Aral, M.; Zencirci, B.; Oksuz, H. Association of maternal serum CRP, IL-6, TNF-alpha, homocysteine, folic acid and vitamin B12 levels with the severity of preeclampsia and fetal birth weight. Hypertens. Pregnancy 2009, 28, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Makedos, G.; Papanicolaou, A.; Hitoglou, A.; Kalogiannidis, I.; Makedos, A.; Vrazioti, V.; Goutzioulis, M. Homocysteine, folic acid and B12 serum levels in pregnancy complicated with preeclampsia. Arch. Gynecol. Obstet. 2007, 275, 121–124. [Google Scholar] [CrossRef]
- Patrick, T.E.; Powers, R.W.; Daftary, A.R.; Ness, R.B.; Roberts, J.M. Homocysteine and folic acid are inversely related in black women with preeclampsia. Hypertension 2004, 43, 1279–1282. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.E.; Zhang, C.; Rene Malinow, M.; Ware-Jauregui, S.; Larrabure, G.; Williams, M.A. Plasma folate, vitamin B(12), and homocyst(e)ine concentrations in preeclamptic and normotensive Peruvian women. Am. J. Epidemiol. 2001, 153, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, N.S.; Patil, V.V.; Mehendale, S.S.; Wagh, G.N.; Gupte, S.A.; Joshi, S.R. Increased homocysteine levels exist in women with preeclampsia from early pregnancy. J. Matern. Fetal. Neonatal. Med. 2016, 29, 2719–2725. [Google Scholar] [CrossRef] [PubMed]
- Dodds, L.; Fell, D.B.; Dooley, K.C.; Armson, B.A.; Allen, A.C.; Nassar, B.A.; Joseph, K.S. Effect of Homocysteine Concentration in Early Pregnancy on Gestational Hypertensive Disorders and Other Pregnancy Outcomes. Clin. Chem. 2008, 54, 326–334. [Google Scholar] [CrossRef]
- Maged, A.M.; Saad, H.; Meshaal, H.; Salah, E.; Abdelaziz, S.; Omran, E.; Katta, M. Maternal serum homocysteine and uterine artery Doppler as predictors of preeclampsia and poor placentation. Arch. Gynecol. Obstet. 2017, 296, 475–482. [Google Scholar] [CrossRef]
- Cotter, A.M.; Molloy, A.M.; Scott, J.M.; Daly, S.F. Elevated plasma homocysteine in early pregnancy: A risk factor for the development of severe preeclampsia. Am. J. Obstet. Gynecol. 2001, 185, 781–785. [Google Scholar] [CrossRef]
- Cotter, A.M.; Molloy, M.; Scott, J.M.; Daly, S. Elevated plasma homocysteine in early pregnancy: A risk factor for the development of nonsevere preeclampsia. Am. J. Obstet. Gynecol. 2003, 189, 391–396. [Google Scholar] [CrossRef]
- Raijmakers, M.T.; Zusterzeel, P.L.; Steegers, E.A.; Peters, W.H. Hyperhomocysteinaemia: A risk factor for preeclampsia? Eur. J. Obstet. Gynecol. Reprod. Biol. 2001, 95, 226–228. [Google Scholar] [CrossRef]
- Hietala, R.; Turpeinen, U.; Laatikainen, T. Serum homocysteine at 16 weeks and subsequent preeclampsia. Obstet. Gynecol. 2001, 97, 527–529. [Google Scholar] [PubMed]
- Hogg, B.B.; Tamura, T.; Johnston, K.E.; Dubard, M.B.; Goldenberg, R.L. Second-trimester plasma homocysteine levels and pregnancy-induced hypertension, preeclampsia, and intrauterine growth restriction. Am. J. Obstet. Gynecol. 2000, 183, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, N.; Mohammad Jafari, R.; Haghnia, S. The evaluation of serum homocysteine, folic acid, and vitamin B12 in patients complicated with preeclampsia. Electron. Physician 2016, 8, 3057–3061. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stuhlinger, M.C.; Oka, R.K.; Graf, E.E.; Schmölzer, I.; Upson, B.M.; Kapoor, O.; Szuba, A.; Malinow, M.R.; Wascher, T.C.; Pachinger, O.; et al. Endothelial dysfunction induced by hyperhomocyst(e)inemia: Role of asymmetric dimethylarginine. Circulation 2003, 108, 933–938. [Google Scholar] [CrossRef]
- Ray, J.G.; Laskin, C.A. Folic acid and homocysteine metabolic defects and the risk of placental abruption, preeclampsia and spontaneous pregnancy loss: A systematic review. Placenta 1999, 20, 519–529. [Google Scholar] [CrossRef]
- Lentz, S.R. Mechanisms of homocysteinee-induced atherothrombosis. J. Thromb. Haemost. 2005, 3, 1646–1654. [Google Scholar] [CrossRef]
- Mao, D.; Che, J.; Li, K.; Han, S.; Yue, Q.; Zhu, L.; Li, L. Association of homocysteine, asymmetric dimethylarginine, and nitric oxide with preeclampsia. Arch. Gynecol. Obstet. 2010, 282, 371–375. [Google Scholar] [CrossRef]
- Powers, R.; Evans, R.; Majors, A.; Ojimba, J.; Ness, R.; Crombleholme, W.R.; Roberts, J.M. Plasma homocysteine concentration is increased in preeclampsia and is associated with evidence of endothelial activation. Am. J. Obstet. Gynecol. 1998, 179, 1605–1611. [Google Scholar] [CrossRef]
- Wollesen, F.; Brattstrom, L.; Refsum, H.; Ueland, P.M.; Berglund, L.; Berne, C. Plasma total homocysteine and cysteine in relation to glomerular filtration rate in diabetes mellitus. Kidney Int. 1999, 55, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Veldman, B.A.; Vervoort, G.; Blom, H.; Smits, P. Reduced plasma total homocysteine concentrations in Type 1 diabetes mellitus is determined by increased renal clearance. Diabet. Med. 2005, 22, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.G.; Lathrop, L. Hyperhomocysteinemia in end-stage renal disease: Prevalence, etiology, and potential relationship to arteriosclerotic outcomes. Kidney Int. 1997, 52, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Maul, H.; Longo, M.; Saade, G.R.; Garfield, R.E. Nitric oxide and its role duringpregnancy: From ovulation to delivery. Curr. Pharm. Des. 2003, 9, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Luzi, G.; Caserta, G.; Iammarino, G.; Clerici, G.; Di Renzo, G.C. Nitric oxide donors in pregnancy: Fetomaternal hemodynamic effects induced in mild pre-eclampsia and threatened preterm labor. Ultrasound Obstetrics Gynecol. 1999, 14, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Ledingham, M.-A.; Denison, F.C.; Kelly, R.W.; Young, A.; Norman, J.E. Nitric oxide donors stimulate prostaglandin F2α and inhibit thromboxane B2 production in the human cervix during the first trimester of pregnancy. Mol. Hum. Reprod. 1999, 5, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A., Jr.; Goncalves, L.F.; Trapani, T.F.; Vieira, S.; Pires, M.; Pires, M.M. Perinatal and hemodynamic evaluation of Sildenafil citrate for preeclampsia treatment: A randomized controlled trial. Obstet. Gynecol. 2016, 128, 253e9. [Google Scholar] [CrossRef] [PubMed]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Longo, M.; Piccinini, F.; Neri, I.; Volpe, A. L-arginine infusion reduces blood pressure in preeclamptic women through nitric oxide release. J. Soc. Gynecol. Investig. 1999, 6, 202–207. [Google Scholar]
- Vadillo-Ortega, F.; Perichart-Perera, O.; Espino, S.; Avila-Vergara, M.A.; Ibarra, I.; Ahued, R.; Strauss, J.F. Effect of supplementation during pregnancy with L-arginine and antioxidant vitamins in medical food on pre-eclampsia in high risk population: Randomised controlled trial. BMJ 2011, 342, d2901. [Google Scholar] [CrossRef]
- Fu, Z.; Ma, Z.; Liu, G.; Wang, L.; Guo, Y. Vitamins supplementation affects the onset of preeclampsia. J. Formos. Med. Assoc. 2018, 117, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Neri, I.; Monari, F.; Sgarbi, L.; Berardi, A.; Masellis, G.; Facchinetti, F. L-arginine supplementation in women with chronic hypertension: Impact on blood pressure and maternal and neonatal complications. J. Matern. Fetal. Neonatal. Med. 2010, 23, 1456–1460. [Google Scholar] [CrossRef] [PubMed]
- Meher, S.; Duley, L. Nitric oxide for preventing pre-eclampsia and its complications. Cochrane Database Syst. Rev. 2007, 2, CD006490. [Google Scholar] [CrossRef] [PubMed]
- Camarena Pulido, E.E.; García Benavides, L.; Panduro Baron, J.G.; Pascoe Gonzalez, S.; Madrigal Saray, A.J.; García Padilla, F.E.; Totsuka Sutto, S.E. Efficacy of L-arginine for preventing preeclampsia in high risk pregnancies: A double-blind randomized clinical trial. Hypertens. Pregnancy 2016, 35, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Abdelrazik, M.; ElBerry, S.; Abosereah, M.; Edris, Y.; Sharafeldeen, A. Prophylactic treatment for preeclampsia in high risk teenage primigravida with nitricoxide donors: A pilot study. J. Matern. Fetal. Neonatal. Med. 2016, 29, 2617e20. [Google Scholar]
- Coles, L.T.; Clifton, P.M. Effect of beetroot juice on lowering blood pressure in freeliving, disease-free adults: A randomized, placebo-controlled trial. Nutr. J. 2012, 11, 106. [Google Scholar] [CrossRef]
- Ormesher, L.; Myers, J.E.; Chmiel, C.; Wareing, M.; Greenwood, S.L.; Tropea, T.; Cottrell, E.C. Effects of dietary nitrate supplementation, from beetroot juice, on blood pressure in hypertensive pregnant women: A randomised, double-blind, placebo-controlled feasibility trial. Nitric Oxide 2018, 80, 37–44. [Google Scholar] [CrossRef]
- Moreland, R.B.; Goldstein, I.I.; Kim, N.N.; Traish, A. Sildenafil Citrate, a Selective Phosphodiesterase Type 5 Inhibitor. Trends Endocrinol. Metab. 1999, 10, 97–104. [Google Scholar] [CrossRef]
- Ramesar, S.V.; Mackraj, I.; Gathiram, P.; Moodley, J. Sildenafil citrate decreases sFlt-1 and sEng in pregnant l-NAME treated Sprague-Dawley rats. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 136–140. [Google Scholar] [CrossRef]
- Herraiz, S.; Pellicer, B.; Serra, V.; Cauli, O.; Cortijo, J.; Felipo, V.; Pellicer, A. Sildenafil citrate improves perinatal outcome in fetuses from pre-eclamptic rats. BJOG 2012, 119, 1394–1402. [Google Scholar] [CrossRef]
- Cacciatore, B.; Halmesmäki, E.; Kaaja, R.; Teramo, K.; Ylikorkala, O. Effects of transdermal nitroglycerin on impedance to flow in the uterine, umbilical, and fetal middle cerebral arteries in pregnancies complicated by preeclampsia and intrauterine growth retardation. Am. J. Obstet. Gynecol. 1998, 179, 140–145. [Google Scholar] [CrossRef]
- Trapani, A., Jr.; Gonçalves, L.F.; Pires, M.M. Transdermal nitroglycerin in patients with severe pre-eclampsia with placental insufficiency: Effect on uterine, umbilical and fetal middle cerebral artery resistance indices. Ultrasound Obstet. Gynecol. 2011, 38, 389–394. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dymara-Konopka, W.; Laskowska, M. The Role of Nitric Oxide, ADMA, and Homocysteine in The Etiopathogenesis of Preeclampsia—Review. Int. J. Mol. Sci. 2019, 20, 2757. https://doi.org/10.3390/ijms20112757
Dymara-Konopka W, Laskowska M. The Role of Nitric Oxide, ADMA, and Homocysteine in The Etiopathogenesis of Preeclampsia—Review. International Journal of Molecular Sciences. 2019; 20(11):2757. https://doi.org/10.3390/ijms20112757
Chicago/Turabian StyleDymara-Konopka, Weronika, and Marzena Laskowska. 2019. "The Role of Nitric Oxide, ADMA, and Homocysteine in The Etiopathogenesis of Preeclampsia—Review" International Journal of Molecular Sciences 20, no. 11: 2757. https://doi.org/10.3390/ijms20112757
APA StyleDymara-Konopka, W., & Laskowska, M. (2019). The Role of Nitric Oxide, ADMA, and Homocysteine in The Etiopathogenesis of Preeclampsia—Review. International Journal of Molecular Sciences, 20(11), 2757. https://doi.org/10.3390/ijms20112757