The High Mobility Group A1 (HMGA1) Chromatin Architectural Factor Modulates Nuclear Stiffness in Breast Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Reversion of the Mesenchymal Phenotype in Triple Negative Breast Cancer (TNBC) Cells Causes an Increase in Cellular Stiffness
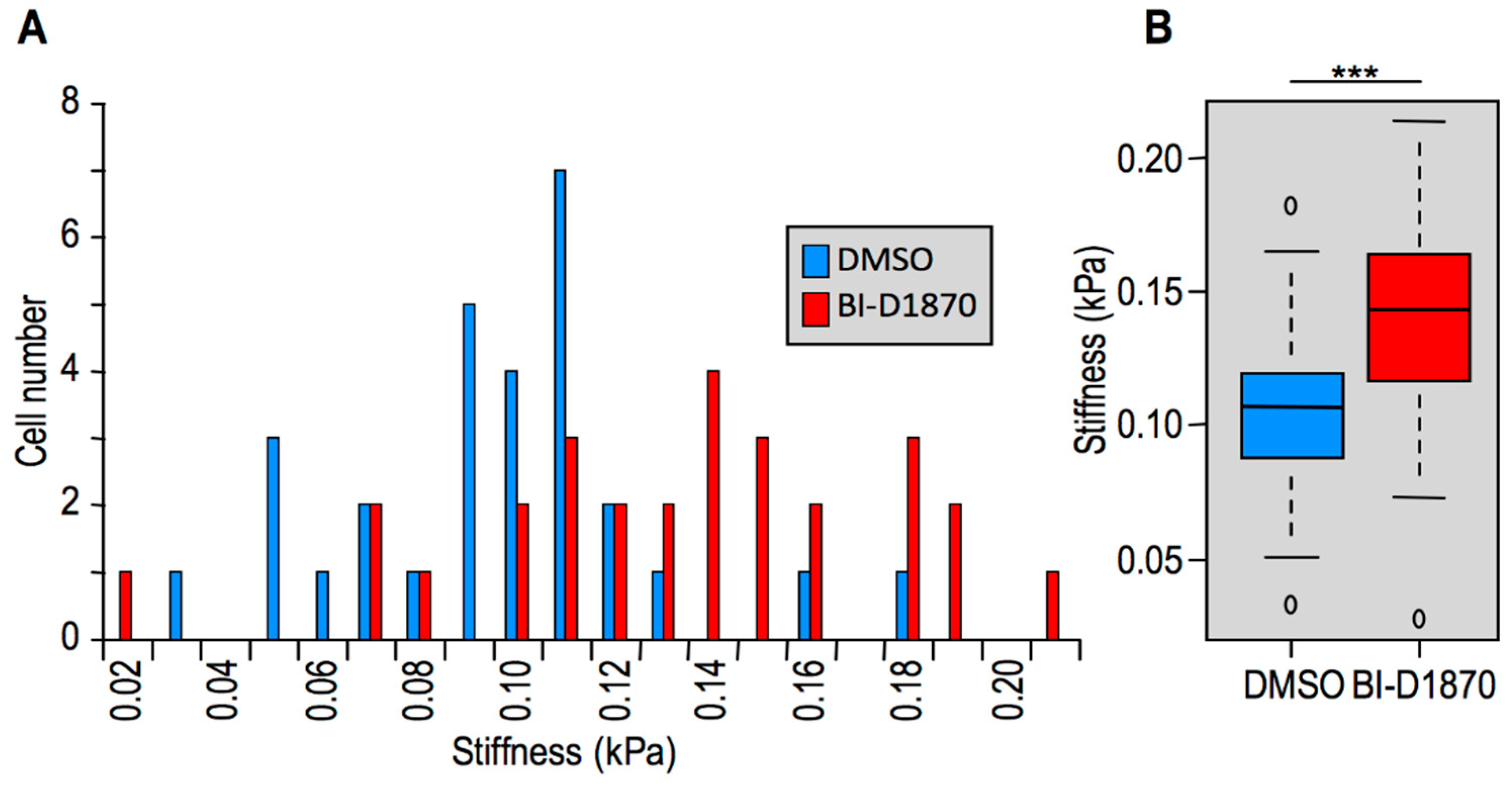
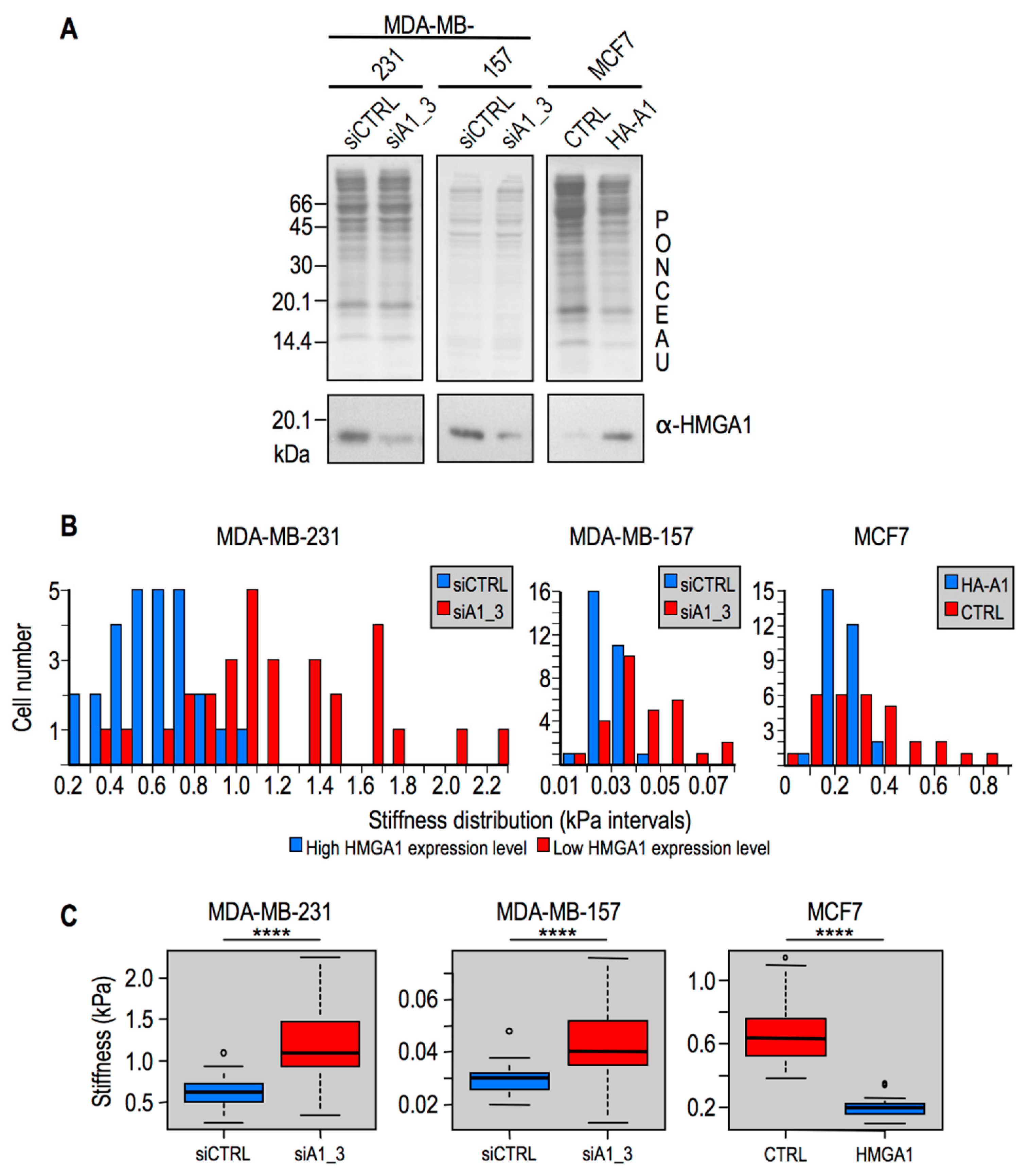
2.2. Modulation of HMGA1 Expression Levels Alters Cellular Stiffness in Breast Cancer Cell Lines
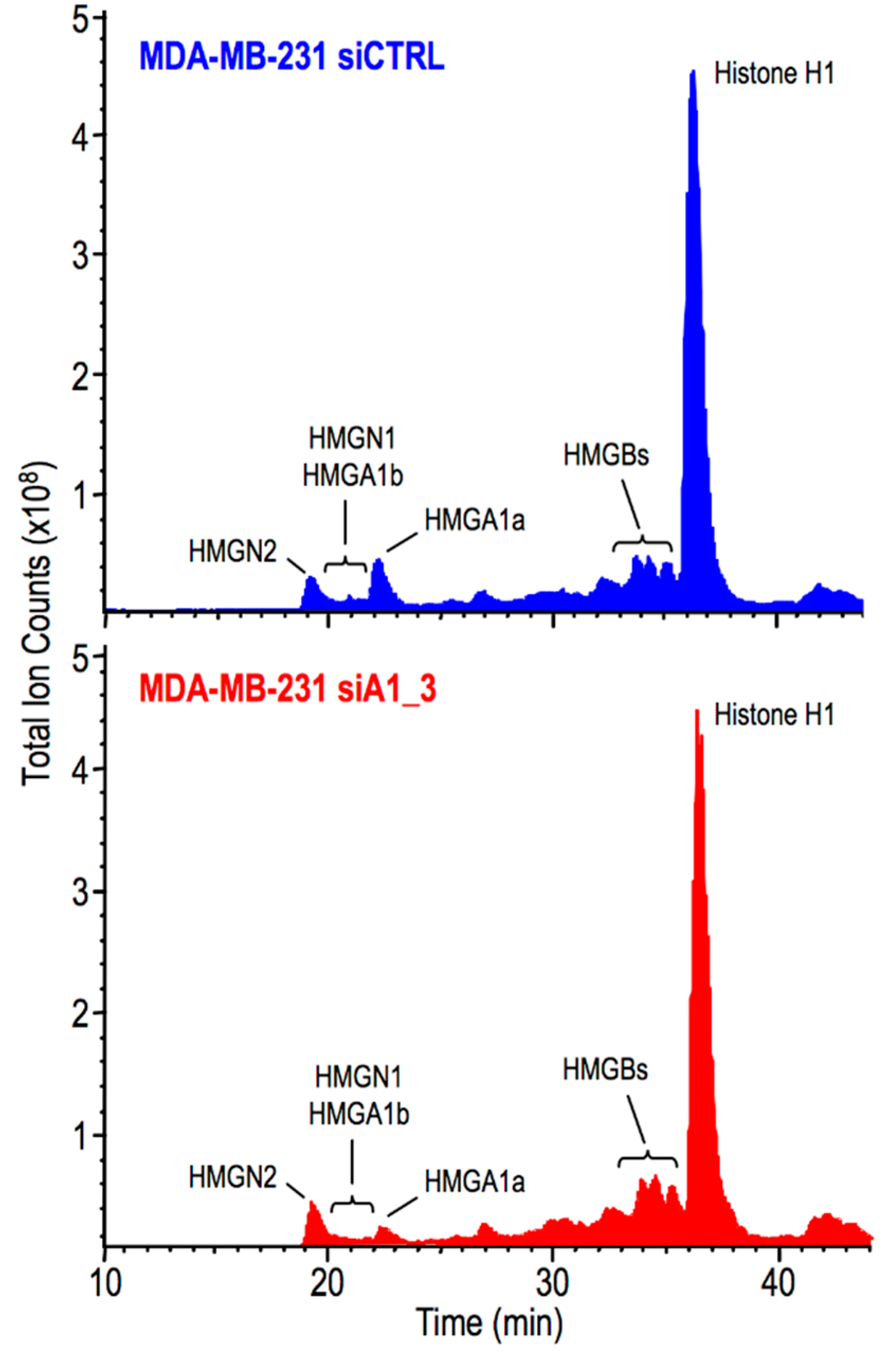
2.3. HMGA1 Expression Is Linked to Histone H1 Phosphorylation Level
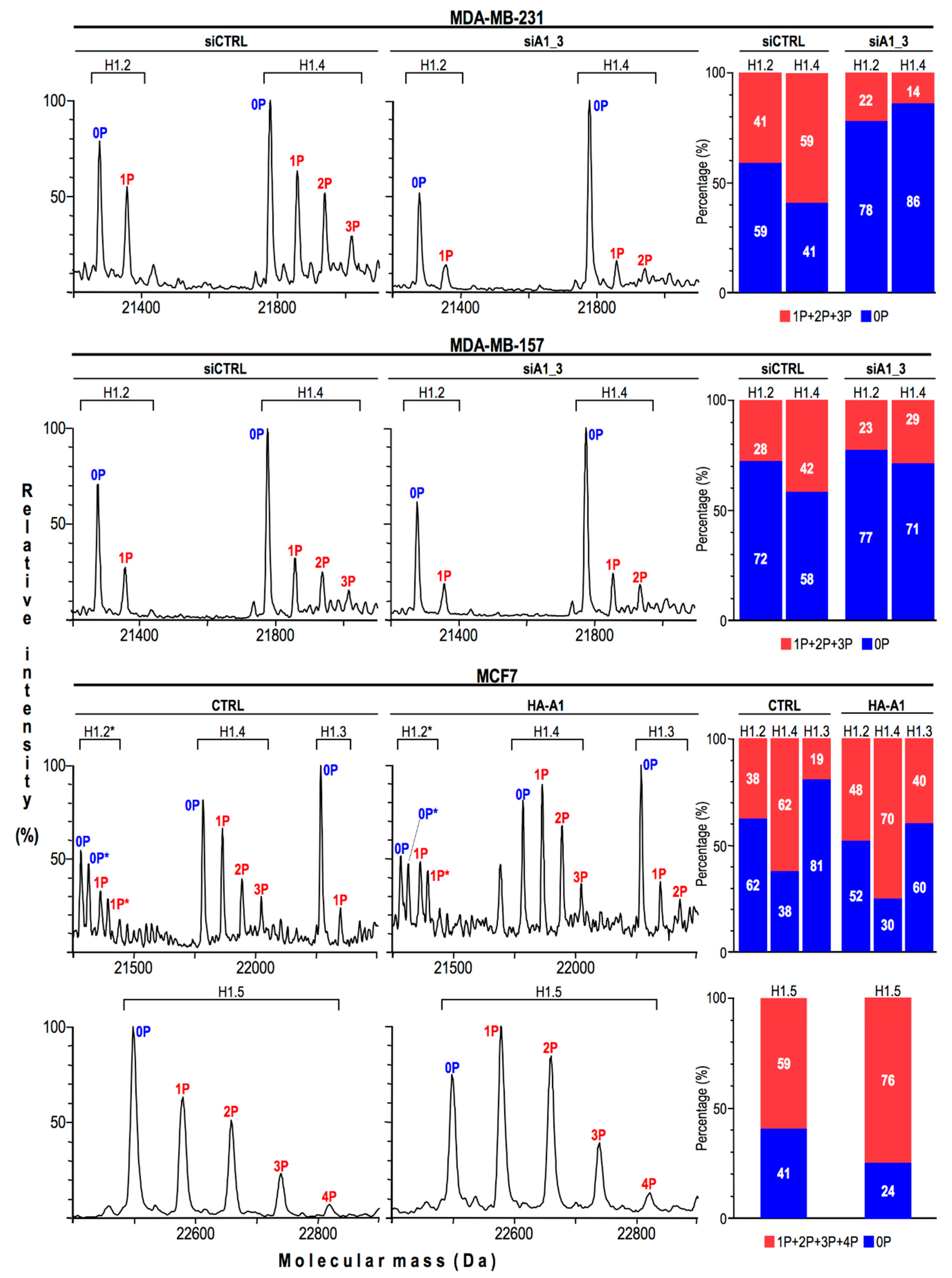
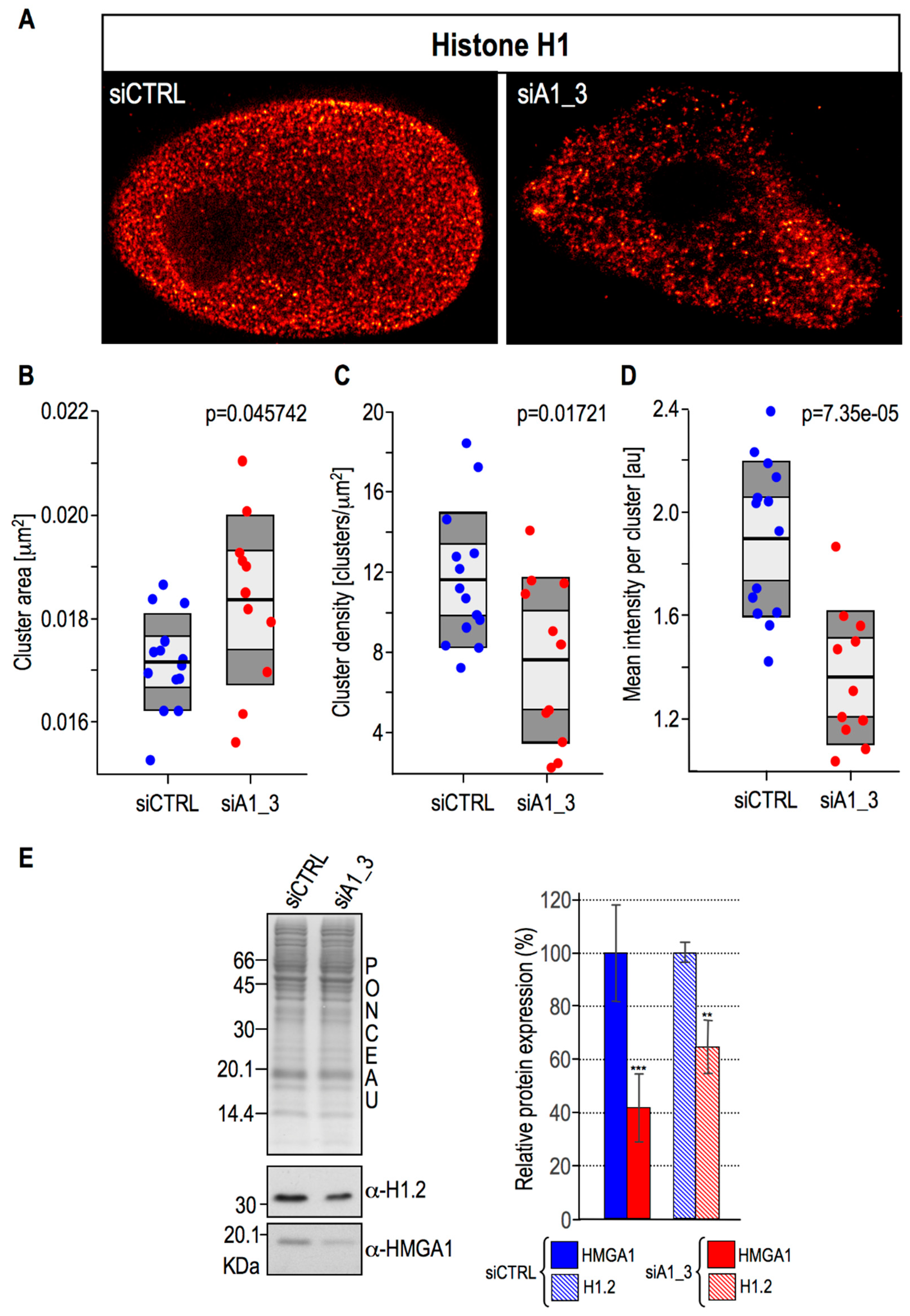
2.4. HMGA1 Expression Is Linked Both to the Spatial Organization within Chromatin and Protein Expression Levels Of Histone H1
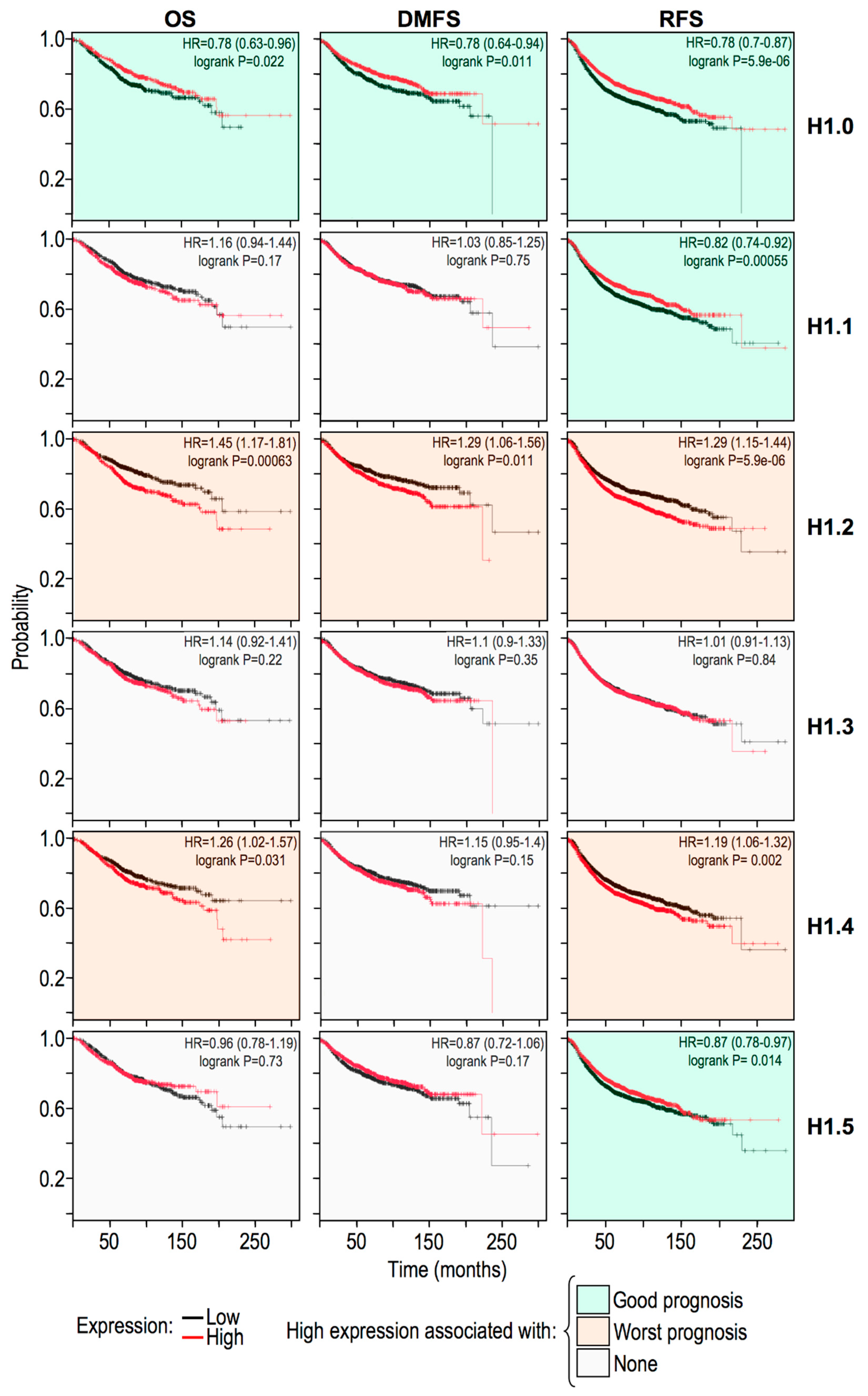
2.5. Histone H1 Variants Have Differential Prognostic Values
3. Discussion
- (1)
- (2)
- HMGA1 and histone H1 exert opposite effects: HMGA1 contributes to chromatin decondensation whereas histone H1 is involved in chromatin compaction [13];
- (3)
- HMGA1 regulates the expression of factors involved in histone H1 phosphorylation (i.e., cyclin E2) [34];
- (4)
- HMGA1 is involved in the recruitment on chromatin of factors involved in histone H1 phosphorylation. HMGA1 binds the 7SK non-coding RNA that in turn is in complex with P-TEFp (i.e., CDK9/cyclinT1/T2) [55];
- (5)
- Phosphorylation decreases histone H1 DNA binding affinity and hence HMGA1 could compete more efficiently with histone H1 [41].
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. SDS PAGE and Western Blot Analyses
4.3. Atomic Force Microscopy
4.4. LC-MS Analyses
4.5. Immunofluorescence Analyses
4.6. Breast Cancer Data Sets and Survival Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ECM | extracellular matrix |
| MMPs | matrix metalloproteinases |
| HMG | High Mobility Group |
| EMT | epithelial-to-mesenchymal transition |
| TNBC | triple negative breast cancer |
| MET | mesenchymal-to-epithelial transition |
| AFM | atomic force microscopy |
| PTMs | post-translational modifications |
| LC–MS | liquid chromatography-mass spectrometry |
| TICs | total ion current chromatograms |
| STORM | stochastic optical reconstruction microscopy |
| TSA | Trichostatin A |
| STED | stimulated emission depletion |
| FRAP | fluorescence recovery after photobleaching |
| MMTV | mouse mammary tumor virus |
| PR | progesterone receptor |
| NF1 | nuclear factor 1 |
References
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Kuang, D.; Zhang, B.; Song, G. Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.M.; Denais, C.; Bakshi, M.C.; Lammerding, J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell. Mol. Bioeng. 2014, 7, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, Y.A.; Nava, M.M.; Wickström, S.A. Emerging roles of mechanical forces in chromatin regulation. J. Cell Sci. 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [PubMed]
- Gerlitz, G.; Bustin, M. The role of chromatin structure in cell migration. Trends Cell Biol. 2011, 21, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Gorjánácz, M. Nuclear assembly as a target for anti-cancer therapies. Nucleus 2014, 5, 47–55. [Google Scholar] [CrossRef]
- Bustin, M.; Misteli, T. Nongenetic functions of the genome. Science 2016, 352, aad6933. [Google Scholar] [CrossRef]
- Stephens, A.D.; Liu, P.Z.; Banigan, E.J.; Almassalha, L.M.; Backman, V.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 2018, 29, 220–233. [Google Scholar] [CrossRef]
- Rochman, M.; Malicet, C.; Bustin, M. HMGN5/NSBP1: A new member of the HMGN protein family that affects chromatin structure and function. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 86–92. [Google Scholar] [CrossRef]
- Furusawa, T.; Rochman, M.; Taher, L.; Dimitriadis, E.K.; Nagashima, K.; Anderson, S.; Bustin, M. Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nat. Commun. 2015, 6, 6138. [Google Scholar] [CrossRef] [PubMed]
- Bustin, M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci. 2001, 263, 152–153. [Google Scholar] [CrossRef]
- Catez, F.; Yang, H.; Tracey, K.J.; Reeves, R.; Misteli, T.; Bustin, M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol. 2004, 24, 4321–4328. [Google Scholar] [CrossRef] [PubMed]
- Rogalla, P.; Drechsler, K.; Frey, G.; Hennig, Y.; Helmke, B.; Bonk, U.; Bullerdiek, J. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am. J. Pathol. 1996, 149, 775–779. [Google Scholar]
- Chiappetta, G.; Avantaggiato, V.; Visconti, R.; Fedele, M.; Battista, S.; Trapasso, F.; Merciai, B.M.; Fidanza, V.; Giancotti, V.; Santoro, M.; et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996, 13, 2439–2446. [Google Scholar]
- Sgarra, R.; Rustighi, A.; Tessari, M.A.; Di Bernardo, J.; Altamura, S.; Fusco, A.; Manfioletti, G.; Giancotti, V. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004, 574, 1–8. [Google Scholar] [CrossRef]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef]
- Maurizio, E.; Cravello, L.; Brady, L.; Spolaore, B.; Arnoldo, L.; Giancotti, V.; Manfioletti, G.; Sgarra, R. Conformational role for the C-terminal tail of the intrinsically disordered high mobility group A (HMGA) chromatin factors. J. Proteome Res. 2011, 10, 3283–3291. [Google Scholar] [CrossRef]
- Pegoraro, S.; Ros, G.; Piazza, S.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef]
- Maurizio, E.; Wiśniewski, J.R.; Ciani, Y.; Amato, A.; Arnoldo, L.; Penzo, C.; Pegoraro, S.; Giancotti, V.; Zambelli, A.; Piazza, S.; et al. Translating Proteomic Into Functional Data: An High Mobility Group A1 (HMGA1) Proteomic Signature Has Prognostic Value in Breast Cancer. Mol. Cell. Proteom. 2016, 15, 109–123. [Google Scholar] [CrossRef]
- Resmini, G.; Rizzo, S.; Franchin, C.; Zanin, R.; Penzo, C.; Pegoraro, S.; Ciani, Y.; Piazza, S.; Arrigoni, G.; Sgarra, R.; et al. HMGA1 regulates the Plasminogen activation system in the secretome of breast cancer cells. Sci. Rep. 2017, 7, 11768. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Cope, L.; Poh, W.; Belton, A.; Roy, S.; Talbot, C.C., Jr.; Sukumar, S.; Huso, D.; Resar, L.M.S. HMGA1: A Master Regulator of Tumor Progression in Triple-Negative Breast Cancer Cells. PLoS ONE 2013, 8, e63419. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Dement, G.A.; Adair, J.E.; Britt, R.L.; Nie, R.; Shima, J.; Taylor, W.E.; Reeves, R. Human KIT ligand promoter is positively regulated by HMGA1 in breast and ovarian cancer cells. Oncogene 2004, 23, 8557–8562. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sgarra, R.; Zammitti, S.; Lo Sardo, A.; Maurizio, E.; Arnoldo, L.; Pegoraro, S.; Giancotti, V.; Manfioletti, G. HMGA molecular network: From transcriptional regulation to chromatin remodeling. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Tessari, M.A.; Di Bernardo, J.; Rustighi, A.; Zago, P.; Liberatori, S.; Armini, A.; Bini, L.; Giancotti, V.; Manfioletti, G. Discovering high mobility group A molecular partners in tumour cells. Proteomics 2005, 5, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Furlan, C.; Zammitti, S.; Lo Sardo, A.; Maurizio, E.; Di Bernardo, J.; Giancotti, V.; Manfioletti, G. Interaction proteomics of the HMGA chromatin architectural factors. Proteomics 2008, 8, 4721–4732. [Google Scholar] [CrossRef]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef]
- Morishita, A.; Zaidi, M.R.; Mitoro, A.; Sankarasharma, D.; Szabolcs, M.; Okada, Y.; D’Armiento, J.; Chada, K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013, 73, 4289–4299. [Google Scholar] [CrossRef]
- Sgarra, R.; Pegoraro, S.; Ros, G.; Penzo, C.; Chiefari, E.; Foti, D.; Brunetti, A.; Manfioletti, G. High Mobility Group A (HMGA) proteins: Molecular instigators of breast cancer onset and progression. Biochim. Biophys. Acta 2018, 1869, 216–229. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Sapkota, G.P.; Cummings, L.; Newell, F.S.; Armstrong, C.; Bain, J.; Frodin, M.; Grauert, M.; Hoffmann, M.; Schnapp, G.; Steegmaier, M.; et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 2007, 401, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Guin, S.; Padhye, S.S.; Zhou, Y.-Q.; Zhang, R.-W.; Wang, M.-H. Ribosomal protein S6 kinase (RSK)-2 as a central effector molecule in RON receptor tyrosine kinase mediated epithelial to mesenchymal transition induced by macrophage-stimulating protein. Mol. Cancer 2011, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Stratford, A.L.; Reipas, K.; Hu, K.; Fotovati, A.; Brough, R.; Frankum, J.; Takhar, M.; Watson, P.; Ashworth, A.; Lord, C.J.; et al. Targeting p90 ribosomal S6 kinase eliminates tumor-initiating cells by inactivating Y-box binding protein-1 in triple-negative breast cancers. Stem Cells 2012, 30, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Ros, G.; Ciani, Y.; Sgarra, R.; Piazza, S.; Manfioletti, G. A novel HMGA1-CCNE2-YAP axis regulates breast cancer aggressiveness. Oncotarget 2015, 6, 19087–19101. [Google Scholar] [CrossRef] [PubMed]
- Pellarin, I.; Arnoldo, L.; Costantini, S.; Pegoraro, S.; Ros, G.; Penzo, C.; Triolo, G.; Demarchi, F.; Sgarra, R.; Vindigni, A.; et al. The architectural chromatin factor high mobility group A1 enhances dna Ligase IV activity influencingdna repair. PLoS ONE 2016, 11, e0164258. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Edberg, D.D.; Li, Y. Architectural Transcription Factor HMGI(Y) Promotes Tumor Progression and Mesenchymal Transition of Human Epithelial Cells. Mol. Cell. Biol. 2001, 21, 575–594. [Google Scholar] [CrossRef] [PubMed]
- Chalut, K.J.; Höpfler, M.; Lautenschläger, F.; Boyde, L.; Chan, C.J.; Ekpenyong, A.; Martinez-Arias, A.; Guck, J. Chromatin decondensation and nuclear softening accompany Nanog downregulation in embryonic stem cells. Biophys. J. 2012, 103, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Harrer, M.; Lührs, H.; Bustin, M.; Scheer, U.; Hock, R. Dynamic interaction of HMGA1a proteins with chromatin. J. Cell Sci. 2004, 117, 3459–3471. [Google Scholar] [CrossRef]
- Hergeth, S.P.; Schneider, R. The H1 linker histones: Multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef]
- Postnikov, Y.V.; Bustin, M. Functional interplay between histone H1 and HMG proteins in chromatin. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 462–467. [Google Scholar] [CrossRef]
- Liao, R.; Mizzen, C.A. Interphase H1 phosphorylation: Regulation and functions in chromatin. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Maurizio, E.; Zammitti, S.; Lo Sardo, A.; Giancotti, V.; Manfioletti, G. Macroscopic differences in HMGA oncoproteins post-translational modifications: C-terminal phosphorylation of HMGA2 affects its DNA binding properties. J. Proteome Res. 2009, 8, 2978–2989. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Celona, B.; Weiner, A.; Di Felice, F.; Mancuso, F.M.; Cesarini, E.; Rossi, R.L.; Gregory, L.; Baban, D.; Rossetti, G.; Grianti, P.; et al. Substantial Histone reduction modulates Genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011, 9, e1001086. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Pedulla, M.L.; Reeves, R. Directional binding of HMG-I(Y) on four-way junction DNA and the molecular basis for competitive binding with HMG-1 and histone H1. Nucleic Acids Res. 1999, 27, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, E.; Bandu, M.T.; Doly, J. Specific binding of high-mobility-group I (HMGI) protein and histone H1 to the upstream AT-rich region of the murine beta interferon promoter: HMGI protein acts as a potential antirepressor of the promoter. Mol. Cell. Biol. 1999, 19, 2803–2816. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Reeves, R. Competition between HMG-I(Y), HMG-1 and histone H1 on four-way junction DNA. Nucleic Acids Res. 1997, 25, 3523–3531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Käs, E.; Gonzalez, E.; Laemmli, U.K. SAR-dependent mobilization of histone H1 by HMG-I/Y in vitro: HMG-I/Y is enriched in H1-depleted chromatin. EMBO J. 1993, 12, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Diani, E.; Beato, M.; Jordan, A. Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet. 2008, 4, e1000227. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shao, Z.; Wang, H.; Yang, J.; Zhang, F.; Luo, Y.; Xu, L.; Ding, Y.; Zhao, L. LASP1-S100A11 axis promotes colorectal cancer aggressiveness by modulating TGFβ/Smad signaling. Sci. Rep. 2016, 6, 26112. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Takahashi, S.; Asamoto, M.; Nakanishi, M.; Wakita, T.; Ogura, Y.; Yatabe, Y.; Shirai, T. Histone H1 expression in human prostate cancer tissues and cell lines. Pathol. Int. 2012, 62, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, C.M.; Schiera, G.; Di Liegro, I. H1.0 Linker Histone as an Epigenetic Regulator of Cell Proliferation and Differentiation. Genes 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Lyubitelev, A.V.; Nikitin, D.V.; Shaytan, A.K.; Studitsky, V.M.; Kirpichnikov, M.P. Structure and Functions of Linker Histones. Biochemistry (Mosc.) 2016, 81, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Eilebrecht, S.; Benecke, B.J.; Benecke, A. 7SK snRNA-mediated, gene-specific cooperativity of HMGA1 and P-TEFb. RNA Biol. 2011, 8, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Fujii, Y.; Hirabayashi, Y.; Gotoh, Y. HMGA regulates the global chromatin state and neurogenic potential in neocortical precursor cells. Nat. Neurosci. 2012, 15, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.C.-N.; Wong, G.L.-H.; Chan, A.W.-H.; Wong, V.W.-S.; Fong, A.K.-W.; Cheung, Y.-S.; Wong, J.; Lee, K.-F.; Chan, S.L.; Lai, P.B.-S.; et al. Liver stiffness measurement predicts high-grade post-hepatectomy liver failure: A prospective cohort study. J. Gastroenterol. Hepatol. 2017, 32, 506–514. [Google Scholar] [CrossRef]
- Zhao, X.; Zhong, Y.; Ye, T.; Wang, D.; Mao, B. Discrimination Between Cervical Cancer Cells and Normal Cervical Cells Based on Longitudinal Elasticity Using Atomic Force Microscopy. Nanoscale Res. Lett. 2015, 10, 1–8. [Google Scholar] [CrossRef]
- Liao, R.; Mizzen, C.A. Site-specific regulation of histone H1 phosphorylation in pluripotent cell differentiation. Epigenetics Chromatin 2017, 10, 29. [Google Scholar] [CrossRef]
- Belton, A.; Gabrovsky, A.; Bae, Y.K.; Reeves, R.; Iacobuzio-Donahue, C.; Huso, D.L.; Resar, L.M.S. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS ONE 2012, 7, e30034. [Google Scholar] [CrossRef]
- Koop, R.; Di Croce, L.; Beato, M. Histone H1 enhances synergistic activation of the MMTV promoter in chromatin. EMBO J. 2003, 22, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.L.; Goldmann, W.H. Feeling the forces: Atomic force microscopy in cell biology. Life Sci. 2003, 72, 2553–2560. [Google Scholar] [CrossRef]
- Sneddon, I.N. The relation between load and penetration in the axisymmetric boussinesq problem for a punch of arbitrary profile. Int. J. Eng. Sci. 1965, 3, 47–57. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Collinet, C.; Stöter, M.; Bradshaw, C.R.; Samusik, N.; Rink, J.C.; Kenski, D.; Habermann, B.; Buchholz, F.; Henschel, R.; Mueller, M.S.; et al. Systems survey of endocytosis by multiparametric image analysis. Nature 2010, 464, 243–249. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.H.; Jahnel, M.; Lauer, J.; Avellaneda, M.J.; Brouilly, N.; Cezanne, A.; Morales-Navarrete, H.; Perini, E.D.; Ferguson, C.; Lupas, A.N.; et al. An endosomal tether undergoes an entropic collapse to bring vesicles together. Nature 2016, 537, 107–111. [Google Scholar] [CrossRef]
- Nagy, A.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Ringnér, M.; Fredlund, E.; Häkkinen, J.; Borg, Å.; Staaf, J. GOBO: Gene expression-based outcome for breast cancer online. PLoS ONE 2011, 6, e17911. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senigagliesi, B.; Penzo, C.; Severino, L.U.; Maraspini, R.; Petrosino, S.; Morales-Navarrete, H.; Pobega, E.; Ambrosetti, E.; Parisse, P.; Pegoraro, S.; et al. The High Mobility Group A1 (HMGA1) Chromatin Architectural Factor Modulates Nuclear Stiffness in Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2733. https://doi.org/10.3390/ijms20112733
Senigagliesi B, Penzo C, Severino LU, Maraspini R, Petrosino S, Morales-Navarrete H, Pobega E, Ambrosetti E, Parisse P, Pegoraro S, et al. The High Mobility Group A1 (HMGA1) Chromatin Architectural Factor Modulates Nuclear Stiffness in Breast Cancer Cells. International Journal of Molecular Sciences. 2019; 20(11):2733. https://doi.org/10.3390/ijms20112733
Chicago/Turabian StyleSenigagliesi, Beatrice, Carlotta Penzo, Luisa Ulloa Severino, Riccardo Maraspini, Sara Petrosino, Hernan Morales-Navarrete, Enrico Pobega, Elena Ambrosetti, Pietro Parisse, Silvia Pegoraro, and et al. 2019. "The High Mobility Group A1 (HMGA1) Chromatin Architectural Factor Modulates Nuclear Stiffness in Breast Cancer Cells" International Journal of Molecular Sciences 20, no. 11: 2733. https://doi.org/10.3390/ijms20112733
APA StyleSenigagliesi, B., Penzo, C., Severino, L. U., Maraspini, R., Petrosino, S., Morales-Navarrete, H., Pobega, E., Ambrosetti, E., Parisse, P., Pegoraro, S., Manfioletti, G., Casalis, L., & Sgarra, R. (2019). The High Mobility Group A1 (HMGA1) Chromatin Architectural Factor Modulates Nuclear Stiffness in Breast Cancer Cells. International Journal of Molecular Sciences, 20(11), 2733. https://doi.org/10.3390/ijms20112733