Hyperprogression under Immunotherapy



Abstract
1. Introduction
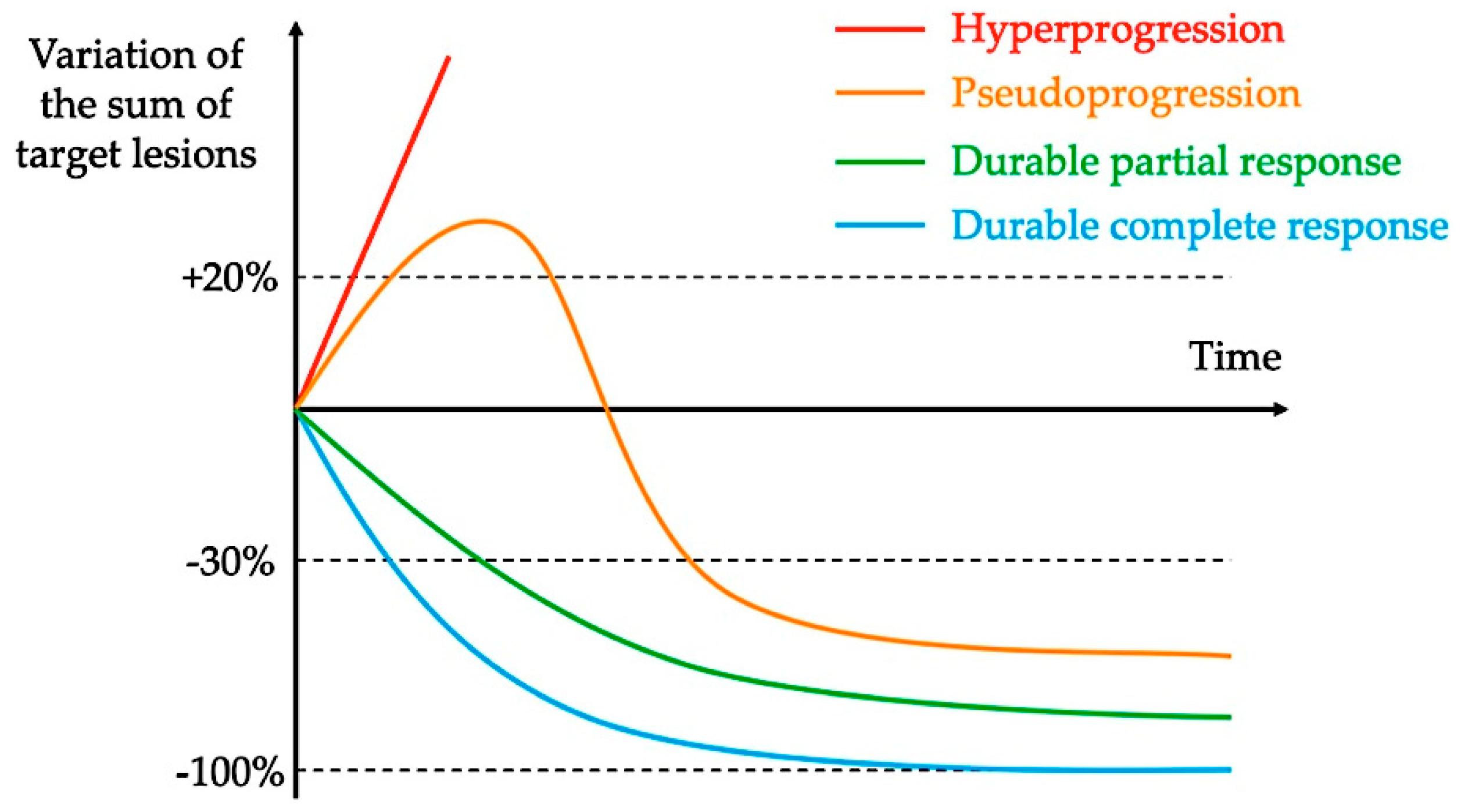
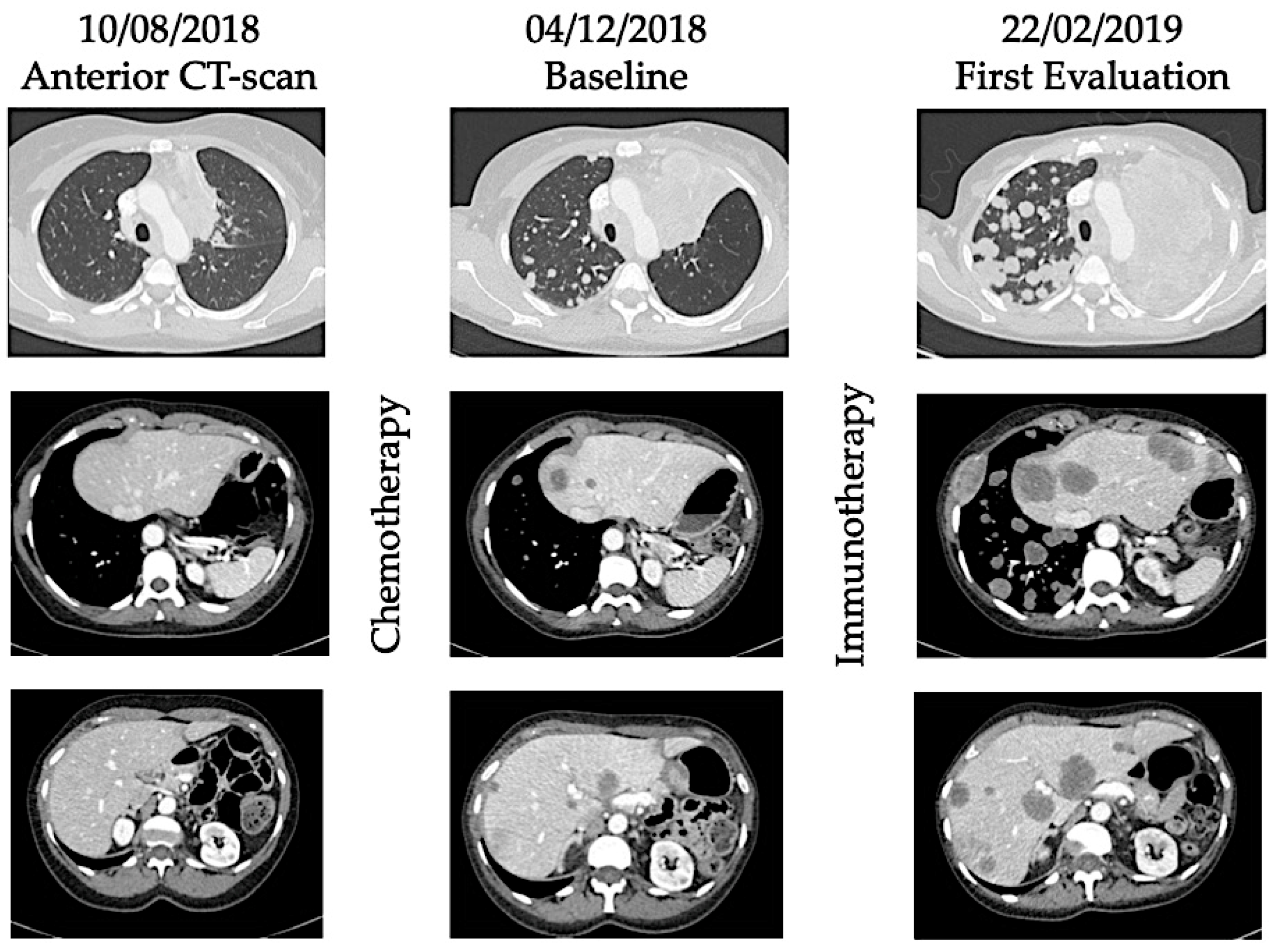
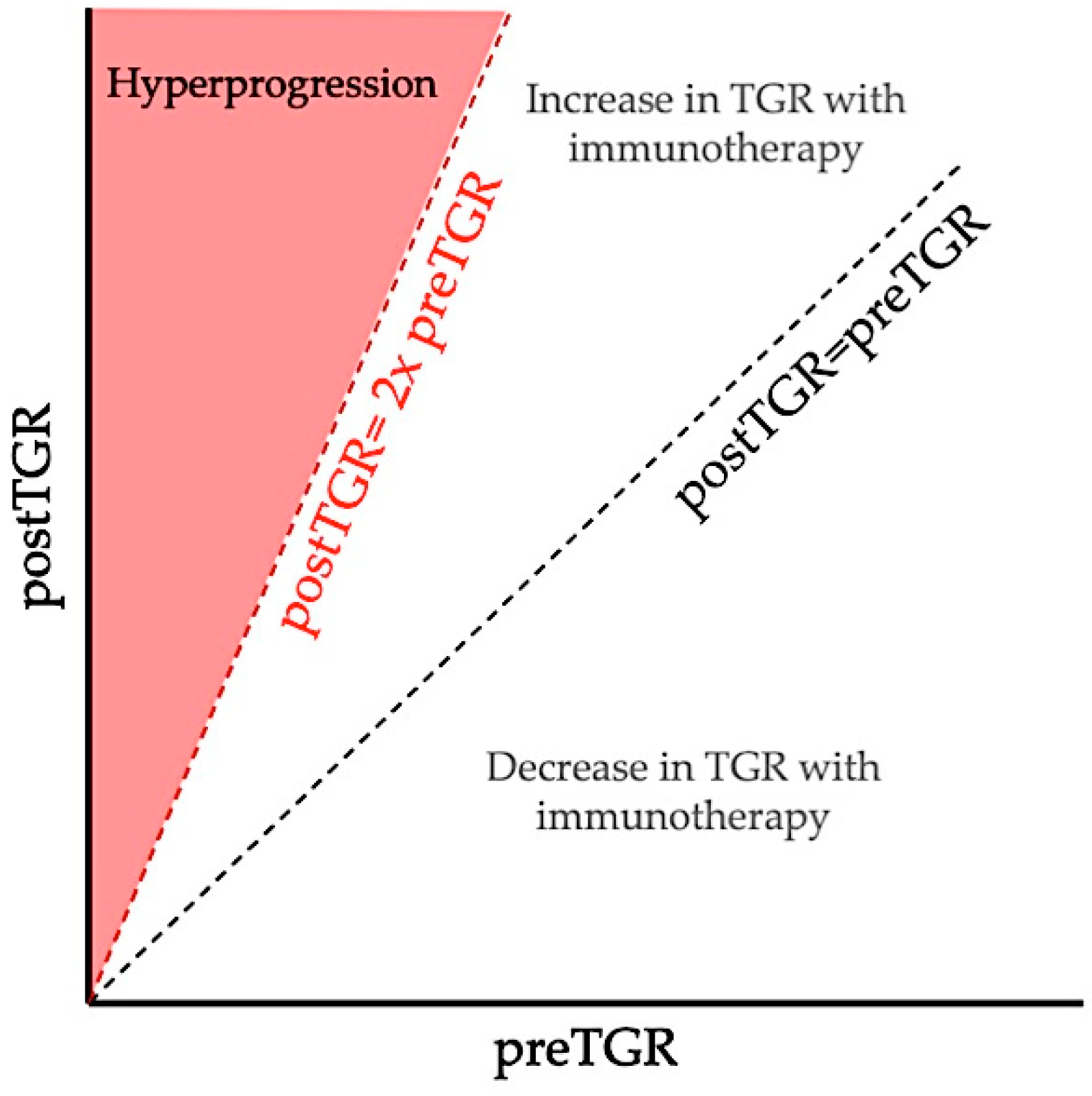
2. Definitions and Reported Incidence of Hyperprogression
3. Predictive Factors
4. Biological Rationale
5. Controversies of the Hyperprogression Phenomenon
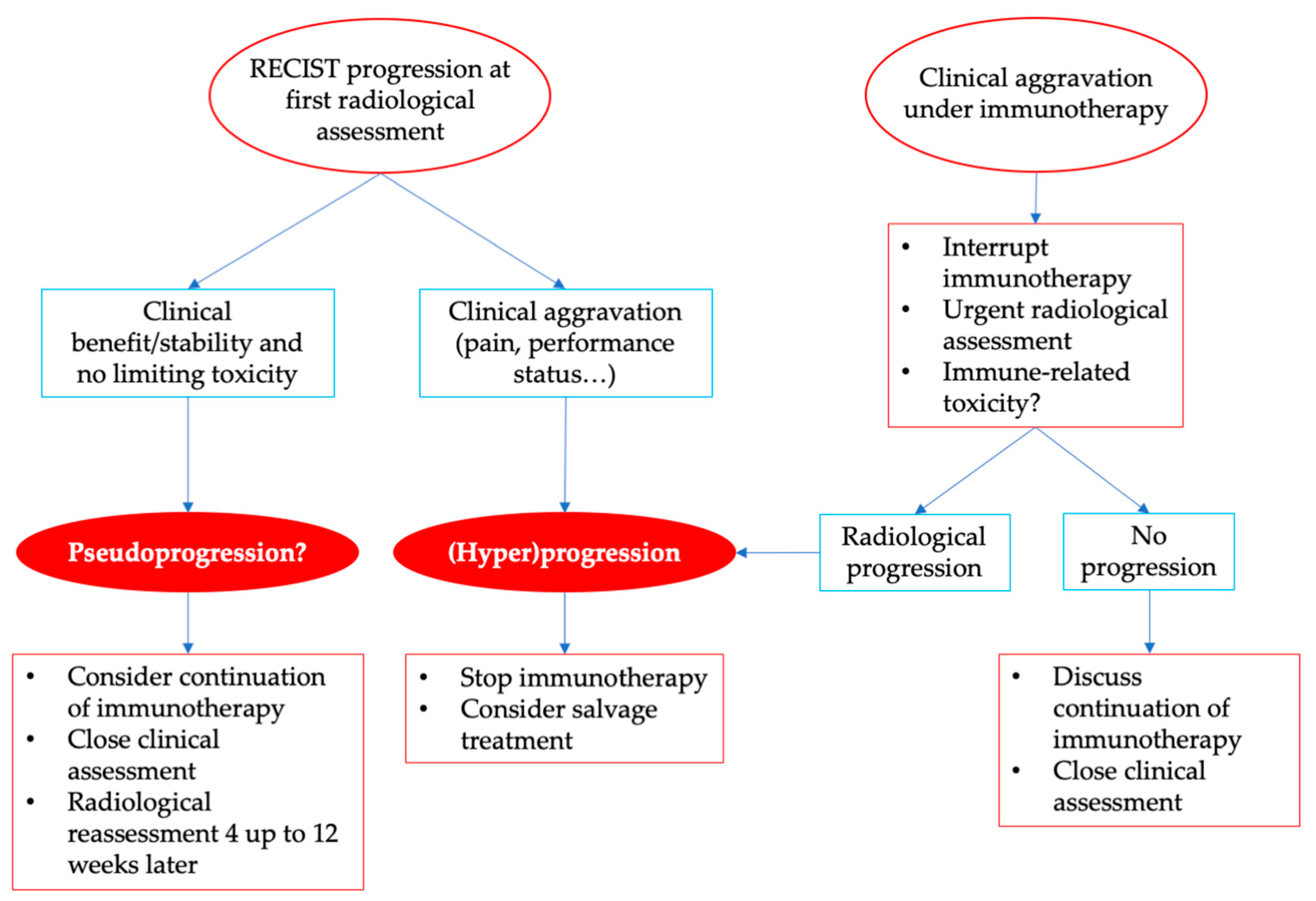
6. Practical Considerations
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| cfDNA | Cell-free DNA |
| CNI | Copy number instability |
| CSC | Cancer stem cells |
| CT scan | Computed-tomography scanner |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| ECOG | Eastern cooperative oncology group |
| Fc | Crystallizable fragment |
| FcR | Crystallizable fragment receptor |
| HNSCC | Head and neck squamous cell carcinoma |
| ICI | Immune checkpoint inhibitor |
| iRECIST | Immune response-evaluation criteria in solid tumors |
| irRC | Immune-related response criteria |
| irRECIST | Immune-related response-evaluation criteria in solid tumors |
| NGS | Next-generation sequencing |
| NSCLC | Non-small-cell lung cancer |
| OS | Overall survival |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PD-L2 | Programmed cell death ligand 2 |
| RECIST | Response-evaluation criteria in solid tumors |
| TAM | Tumor-associated macrophage |
| TGK | Tumor growth kinetics |
| TGR | Tumor growth rate |
| TILs | Tumor-infiltrating T lymphocytes |
| Tregs | Regulatory T-cells |
| TTF | Time to treatment failure |
| uPD | Unconfirmed progressive disease |
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 0, null. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef]
- Nishino, M.; Giobbie-Hurder, A.; Gargano, M.; Suda, M.; Ramaiya, N.H.; Hodi, F.S. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin. Cancer Res. 2013, 19, 3936–3943. [Google Scholar] [CrossRef]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.-C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef]
- Saâda-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611. [Google Scholar] [CrossRef]
- Chubachi, S.; Yasuda, H.; Irie, H.; Fukunaga, K.; Naoki, K.; Soejima, K.; Betsuyaku, T. A Case of Non-Small Cell Lung Cancer with Possible “Disease Flare” on Nivolumab Treatment. Case Rep. Oncol. Med. 2016, 1075641. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef]
- Kanjanapan, Y.; Day, D.; Wang, L.; Al-Sawaihey, H.; Abbas, E.; Namini, A.; Siu, L.L.; Hansen, A.; Razak, A.A.; Spreafico, A.; et al. Hyperprogressive disease in early-phase immunotherapy trials: Clinical predictors and association with immune-related toxicities: Hyperprogressive Disease Immunotherapy. Cancer 2019. [Google Scholar] [CrossRef]
- Russo, G.L.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyper-progressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef]
- Matos, I. Incidence and clinical implications of a new definition of hyperprogression (HPD) with immune checkpoint inhibitors (ICIs) in patients treated in phase 1 (Ph1) trials. J. Clin. Oncol. 2018, 3032. [Google Scholar] [CrossRef]
- Weiss, G.J.; Beck, J.; Braun, D.P.; Bornemann-Kolatzki, K.; Barilla, H.; Cubello, R.; Quan, W.; Sangal, A.; Khemka, V.; Waypa, J.; et al. Tumor Cell-Free DNA Copy Number Instability Predicts Therapeutic Response to Immunotherapy. Clin. Cancer Res. 2017, 23, 5074–5081. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef]
- Kirilovsky, A.; Marliot, F.; El Sissy, C.; Haicheur, N.; Galon, J.; Pagès, F. Rational bases for the use of the Immunoscore in routine clinical settings as a prognostic and predictive biomarker in cancer patients. Int. Immunol. 2016, 28, 373–382. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Ramos, R.N.; Piaggio, E.; Romano, E. Mechanisms of Resistance to Immune Checkpoint Antibodies. Handb. Exp. Pharmacol. 2018, 249, 109–128. [Google Scholar]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.-S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Borcoman, E.; De La Rochere, P.; Richer, W.; Vacher, S.; Chemlali, W.; Krucker, C.; Sirab, N.; Radvanyi, F.; Allory, Y.; Pignot, G.; et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology 2019, 8, e1581556. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Stein, R.G.; Ebert, S.; Schlahsa, L.; Scholz, C.J.; Braun, M.; Hauck, P.; Horn, E.; Monoranu, C.-M.; Thiemann, V.J.; Wustrow, M.P.; et al. Cognate Non-Lytic Interactions between CD8+ T cells and Breast Cancer Cells Induce Cancer Stem Cell-like Properties. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Gil Del Alcazar, C.R.; Huh, S.J.; Ekram, M.B.; Trinh, A.; Liu, L.L.; Beca, F.; Zi, X.; Kwak, M.; Bergholtz, H.; Su, Y.; et al. Immune Escape in Breast Cancer During In Situ to Invasive Carcinoma Transition. Cancer Discov. 2017, 7, 1098–1115. [Google Scholar] [CrossRef]
- Cimino-Mathews, A.; Foote, J.B.; Emens, L.A. Immune targeting in breast cancer. Oncology 2015, 29, 375–385. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Reim, F.; Dombrowski, Y.; Ritter, C.; Buttmann, M.; Häusler, S.; Ossadnik, M.; Krockenberger, M.; Beier, D.; Beier, C.P.; Dietl, J.; et al. Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Res. 2009, 69, 8058–8066. [Google Scholar] [CrossRef]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Kuriyama, Y.; Kim, Y.H.; Nagai, H.; Ozasa, H.; Sakamori, Y.; Mishima, M. Disease flare after discontinuation of crizotinib in anaplastic lymphoma kinase-positive lung cancer. Case Rep. Oncol. 2013, 6, 430–433. [Google Scholar] [CrossRef]
- Chaft, J.E.; Oxnard, G.R.; Sima, C.S.; Kris, M.G.; Miller, V.A.; Riely, G.J. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin. Cancer Res. 2011, 17, 6298–6303. [Google Scholar] [CrossRef]
- Iacovelli, R.; Massari, F.; Albiges, L.; Loriot, Y.; Massard, C.; Fizazi, K.; Escudier, B. Evidence and Clinical Relevance of Tumor Flare in Patients Who Discontinue Tyrosine Kinase Inhibitors for Treatment of Metastatic Renal Cell Carcinoma. Eur. Urol. 2015, 68, 154–160. [Google Scholar] [CrossRef]
- Ogawara, D.; Soda, H.; Iwasaki, K.; Suyama, T.; Taniguchi, H.; Fukuda, Y.; Mukae, H. Remarkable response of nivolumab-refractory lung cancer to salvage chemotherapy. Thorac. Cancer 2018, 9, 175–180. [Google Scholar] [CrossRef]
- Schvartsman, G.; Peng, S.A.; Bis, G.; Lee, J.J.; Benveniste, M.F.K.; Zhang, J.; Roarty, E.B.; Lacerda, L.; Swisher, S.; Heymach, J.V.; et al. Response rates to single-agent chemotherapy after exposure to immune checkpoint inhibitors in advanced non-small cell lung cancer. Lung Cancer 2017, 112, 90–95. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Drugs | Cancer Type | Definition of Hyperprogression | Number of Patients | Rates | Predictive Factors Identified | References |
|---|---|---|---|---|---|---|
| PD-1/PD-L1 inhibitors (phase 1 trials) | All cancer | RECIST progression and ≥2-fold increase TGR | 131 | 9% | Age > 65 years old | [14] |
| PD-1/PD-L1 inhibitors | NSCLC | RECIST progression and ∆TGR > 50% | 406 | 13.8% | >2 metastatic sites | [18] |
| ICI and/or costimulatory molecules (phase 1 trials) | All cancer | RECIST progression and ≥2-fold increase TGR | 182 | 7% | Female gender | [19] |
| PD-1/PD-L1 inhibitors | HNSCC | ≥2-fold increase TGK | 34 | 29% | Regional recurrence in the irradiated field | [15] |
| ICI | NSCLC | RECIST progression and at least 3 of: TTF < 2 months or ≥50% increase of sum of target lesions major diameter or ≥2 new lesions in organ already involved or Spread to a new organ or ECOG PS ≥ 2 | 152 | 25.7% | Density of myeloperoxidase myeloid cells within the tumor Low PD-L1 expression in tumor cells | [20] |
| ICI or costimulatory molecules | All cancer | TTF < 2 months and >50% increase in tumor burden (irRECIST) and ≥2-fold increase in progression pace | 155 | 4% | EGFR, MDM2/4 and DNMT3A alterations | [21] |
| ICI (phase 1 trials) | All cancer | TTF < 2 months and ≥10mm increase in measurable lesions and ≥40% increase in target tumor burden or >20% plus appearance of multiple new lesions | 214 | 15% | [22] |
| RECIST 1.1 [10] | irRC [11] | irRECIST [12] | iRECIST [13] | |
|---|---|---|---|---|
| Lesion measurement | Unidimensional | Bidimensional | Unidimensional | Unidimensional |
| Baseline lesion size | ≥10 mm | 5 × 5 mm | ≥10 mm | ≥10 mm |
| Baseline lesion number | 5 total, 2 per organ | 10 total, 5 per organ | 5 total, 2 per organ | 5 total, 2 per organ |
| CR | Disappearance of all lesions | Disappearance of all lesions | Disappearance of all lesions | Disappearance of all lesions |
| PR | ≥30% decrease from baseline | ≥50% decrease from baseline | ≥30% decrease from baseline | ≥30% decrease from baseline |
| SD | Neither PR or PD | Neither PR or PD | Neither PR or PD | Neither PR or PD |
| PD | ≥20% increase from nadir (≥5 mm) | ≥25% increase from nadir | ≥20% increase from nadir (≥5 mm) | ≥20% increase from nadir (≥5 mm) |
| Confirmed progressive disease | Not applicable | At least 4 weeks after | At least 4 weeks after and up to 12 weeks | At least 4 weeks after and up to 8 weeks |
| Appearance of new lesions | Always PD | Incorporate in the sum of measurement | Incorporate in the sum of measurement | Unconfirmed progressive disease, not included in the sum of measurement |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frelaut, M.; Le Tourneau, C.; Borcoman, E. Hyperprogression under Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2674. https://doi.org/10.3390/ijms20112674
Frelaut M, Le Tourneau C, Borcoman E. Hyperprogression under Immunotherapy. International Journal of Molecular Sciences. 2019; 20(11):2674. https://doi.org/10.3390/ijms20112674
Chicago/Turabian StyleFrelaut, Maxime, Christophe Le Tourneau, and Edith Borcoman. 2019. "Hyperprogression under Immunotherapy" International Journal of Molecular Sciences 20, no. 11: 2674. https://doi.org/10.3390/ijms20112674
APA StyleFrelaut, M., Le Tourneau, C., & Borcoman, E. (2019). Hyperprogression under Immunotherapy. International Journal of Molecular Sciences, 20(11), 2674. https://doi.org/10.3390/ijms20112674