Gastric Leptin and Tumorigenesis: Beyond Obesity

Abstract
1. Introduction
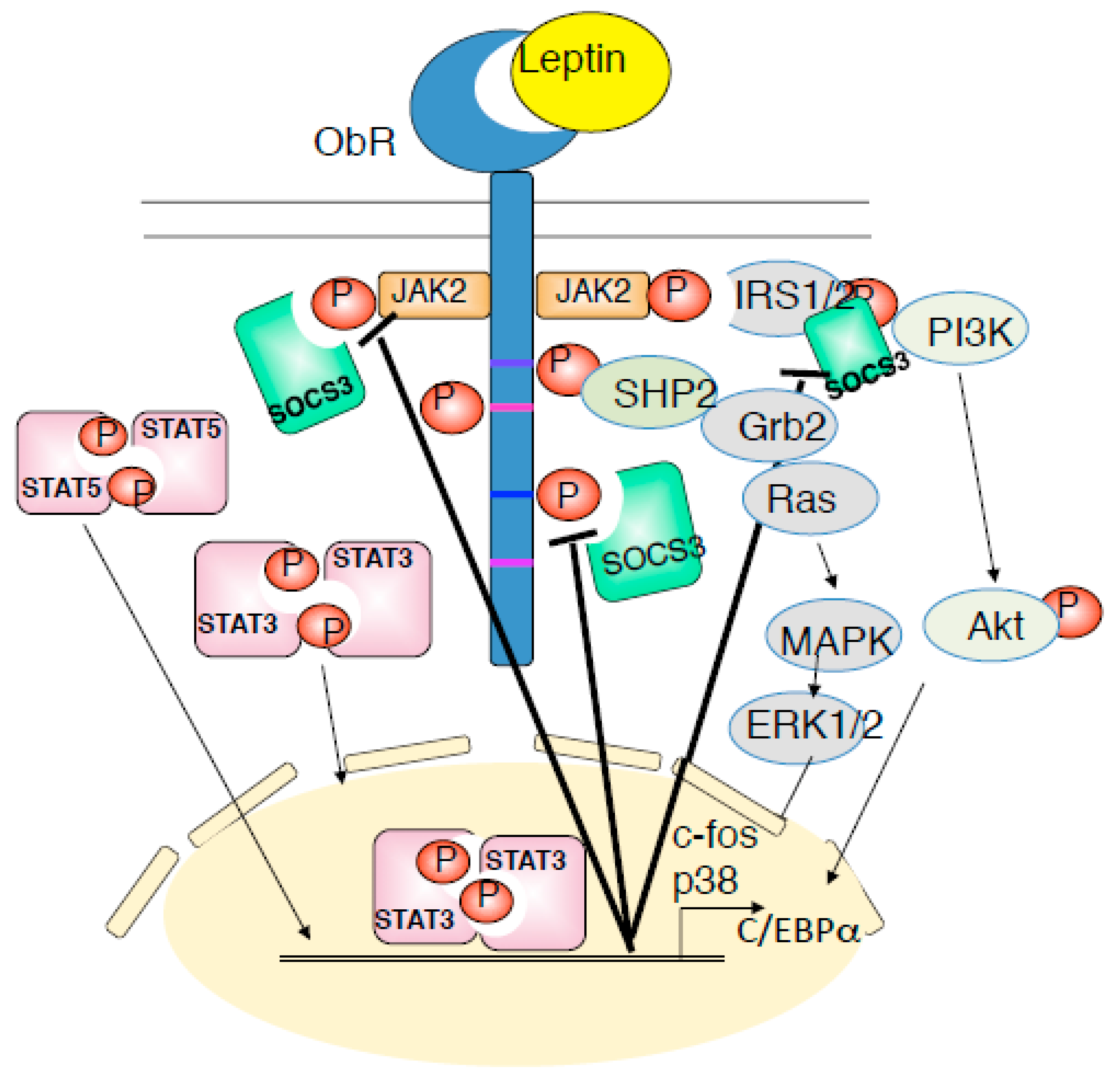
2. Background of Leptin and Its Receptor Signaling
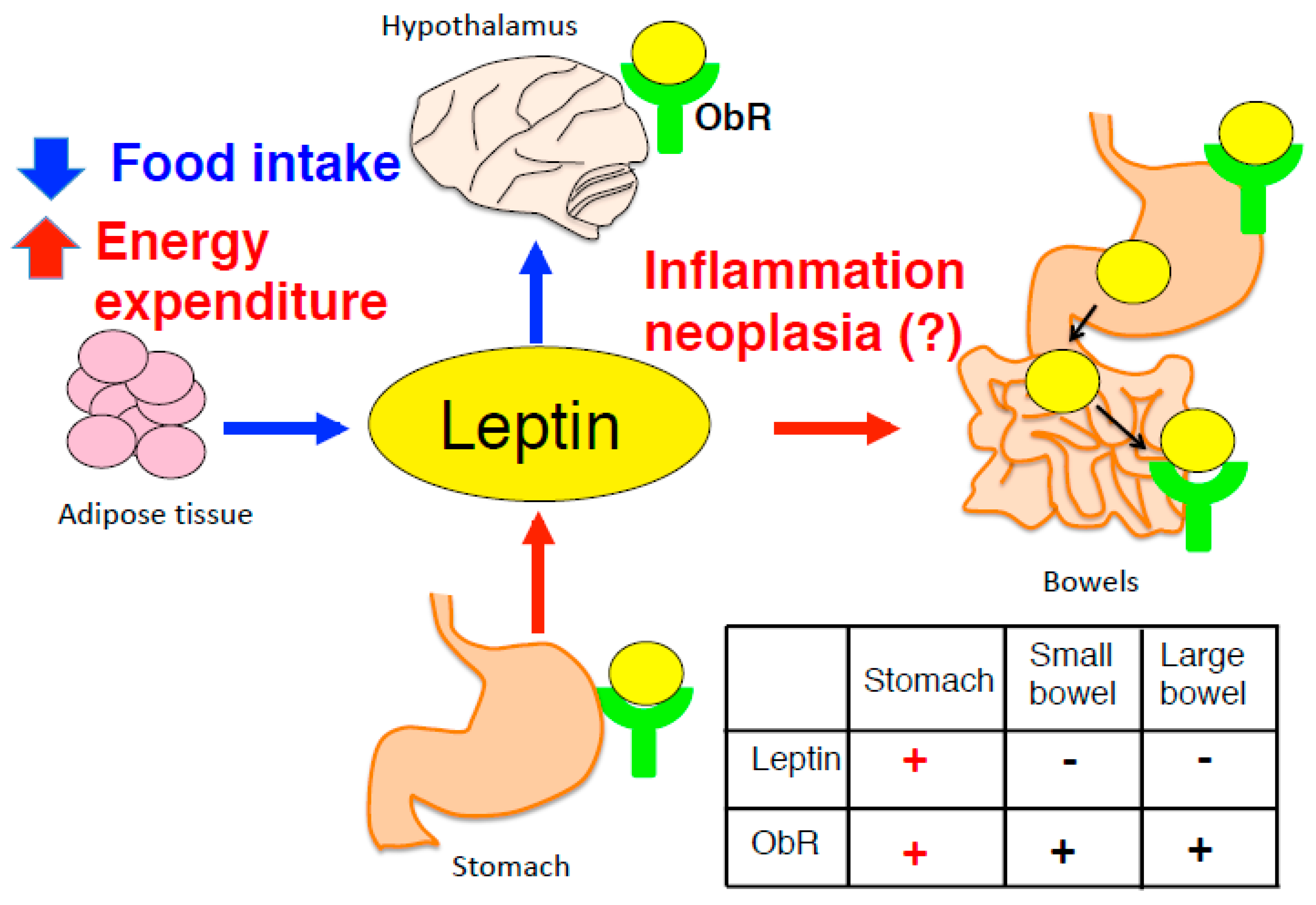
3. Adipocyte-Derived Leptin in the Central Nervous System
4. Gastric Leptin as a Causative Factor of Carcinogenesis in Stomach
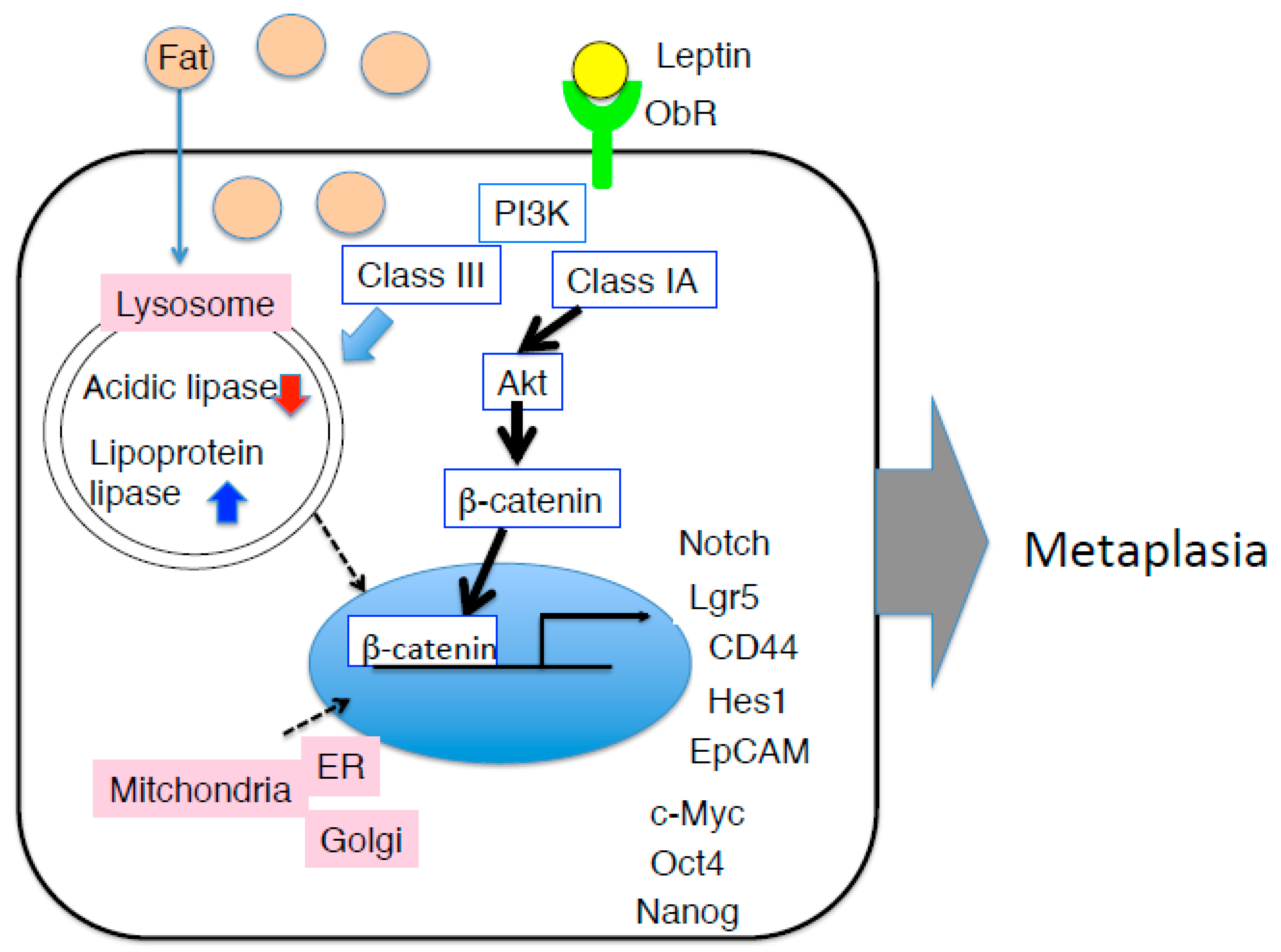
5. Gastric Neoplasia Triggered by Diet-Induced Obesity
6. Therapeutic Implications
7. Conclusions
Funding
Conflicts of Interest
References
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef]
- Bado, A.; Levasseur, S.; Attoub, S.; Kermorgant, S.; Laigneau, J.P.; Bortoluzzi, M.N.; Moizo, L.; Lehy, T.; Guerre-Millo, M.; Le Marchand-Brustel, Y.; et al. The stomach is a source of leptin. Nature 1998, 394, 790–793. [Google Scholar] [CrossRef]
- Masuzaki, H.; Ogawa, Y.; Sagawa, N.; Hosoda, K.; Matsumoto, T.; Mise, H.; Nishimura, H.; Yoshimasa, Y.; Tanaka, I.; Mori, T.; et al. Nonadipose tissue production of leptin: Leptin as a novel placenta-derived hormone in humans. Nat. Med. 1997, 3, 1029–1033. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Ando, S.; Catalano, S. Leptin, obesity and breast cancer: Progress to understanding the molecular connections. Curr. Opin. Pharmacol. 2016, 31, 83–89. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juarez-Cruz, J.C.; Mendoza-Catalan, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling pathways induced by leptin during epithelial(-)mesenchymal transition in breast cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Vannella, L.; Lahner, E.; Annibale, B. Risk for gastric neoplasias in patients with chronic atrophic gastritis: A critical reappraisal. World J. Gastroenterol. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Asombang, A.W.; Ibdah, J.A. Characteristics of gastric cancer in asia. World J. Gastroenterol. 2014, 20, 4483–4490. [Google Scholar] [CrossRef]
- Rivera, F.; Romero, C.; Jimenez-Fonseca, P.; Izquierdo-Manuel, M.; Salud, A.; Martinez, E.; Jorge, M.; Arrazubi, V.; Mendez, J.C.; Garcia-Alfonso, P.; et al. Phase ii study to evaluate the efficacy of trastuzumab in combination with capecitabine and oxaliplatin in first-line treatment of her2-positive advanced gastric cancer: Herxo trial. Cancer Chemother. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Chiariotti, L.; Angrisano, T.; Keller, S.; Florio, E.; Affinito, O.; Pallante, P.; Perrino, C.; Pero, R.; Lembo, F. Epigenetic modifications induced by helicobacter pylori infection through a direct microbe-gastric epithelial cells cross-talk. Med. Microbiol. Immunol. 2013, 202, 327–337. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pero, R.; Coretti, L.; Nigro, E.; Lembo, F.; Laneri, S.; Lombardo, B.; Daniele, A.; Scudiero, O. Beta-defensins in the fight against helicobacter pylori. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, D.H.; Oh, H.S.; Seo, J.Y.; Lee, D.H.; Kim, N.; Jeong, S.H.; Kim, J.W.; Hwang, J.H.; Park, Y.S.; et al. Higher prevalence of obesity in gastric cardia adenocarcinoma compared to gastric non-cardia adenocarcinoma. Dig. Dis. Sci. 2012, 57, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- O’Doherty, M.G.; Freedman, N.D.; Hollenbeck, A.R.; Schatzkin, A.; Abnet, C.C. A prospective cohort study of obesity and risk of oesophageal and gastric adenocarcinoma in the nih-aarp diet and health study. Gut 2012, 61, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.L. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973, 9, 294–298. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morton, G.J.; Stearns, W.H.; Rhodes, C.J.; Myers, M.G., Jr.; Schwartz, M.W. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Al-Qassab, H.; Smith, M.A.; Irvine, E.E.; Guillermet-Guibert, J.; Claret, M.; Choudhury, A.I.; Selman, C.; Piipari, K.; Clements, M.; Lingard, S.; et al. Dominant role of the p110beta isoform of pi3k over p110alpha in energy homeostasis regulation by pomc and agrp neurons. Cell. Metab. 2009, 10, 343–354. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, ob-r. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef]
- Pan, W.; Hsuchou, H.; Tu, H.; Kastin, A.J. Developmental changes of leptin receptors in cerebral microvessels: Unexpected relation to leptin transport. Endocrinology 2008, 149, 877–885. [Google Scholar] [CrossRef]
- Mancour, L.V.; Daghestani, H.N.; Dutta, S.; Westfield, G.H.; Schilling, J.; Oleskie, A.N.; Herbstman, J.F.; Chou, S.Z.; Skiniotis, G. Ligand-induced architecture of the leptin receptor signaling complex. Mol. Cell. 2012, 48, 655–661. [Google Scholar] [CrossRef]
- Howard, J.K.; Flier, J.S. Attenuation of leptin and insulin signaling by socs proteins. Trends Endocrinol. Metab. 2006, 17, 365–371. [Google Scholar] [CrossRef]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Munzberg, H.; Myers, M.G., Jr. The long form of the leptin receptor regulates stat5 and ribosomal protein s6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef]
- Lee, J.Y.; Muenzberg, H.; Gavrilova, O.; Reed, J.A.; Berryman, D.; Villanueva, E.C.; Louis, G.W.; Leinninger, G.M.; Bertuzzi, S.; Seeley, R.J.; et al. Loss of cytokine-stat5 signaling in the cns and pituitary gland alters energy balance and leads to obesity. PLoS ONE 2008, 3, e1639. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Muller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Johren, O.; Pietrzik, C.U.; et al. The lepr-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Elmquist, J.K. Sixteen years and counting: An update on leptin in energy balance. J. Clin. Invest. 2011, 121, 2087–2093. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob rna in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Townsend, K.L.; Lorenzi, M.M.; Widmaier, E.P. High-fat diet-induced changes in body mass and hypothalamic gene expression in wild-type and leptin-deficient mice. Endocrine 2008, 33, 176–188. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Whittington, A.; Barnes, M.J.; Wang, Z.; Bray, G.A.; Morrison, C.D. Hf diets increase hypothalamic ptp1b and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E291–E299. [Google Scholar] [CrossRef]
- Ogus, S.; Ke, Y.; Qiu, J.; Wang, B.; Chehab, F.F. Hyperleptinemia precipitates diet-induced obesity in transgenic mice overexpressing leptin. Endocrinology 2003, 144, 2865–2869. [Google Scholar] [CrossRef]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Wabitsch, M.; Funcke, J.B.; Lennerz, B.; Kuhnle-Krahl, U.; Lahr, G.; Debatin, K.M.; Vatter, P.; Gierschik, P.; Moepps, B.; Fischer-Posovszky, P. Biologically inactive leptin and early-onset extreme obesity. N Engl. J. Med. 2015, 372, 48–54. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of socs-3 as a potential mediator of central leptin resistance. Mol. Cell. 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Mori, H.; Hanada, R.; Hanada, T.; Aki, D.; Mashima, R.; Nishinakamura, H.; Torisu, T.; Chien, K.R.; Yasukawa, H.; Yoshimura, A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004, 10, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kievit, P.; Howard, J.K.; Badman, M.K.; Balthasar, N.; Coppari, R.; Mori, H.; Lee, C.E.; Elmquist, J.K.; Yoshimura, A.; Flier, J.S. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in pomc-expressing cells. Cell. Metab. 2006, 4, 123–132. [Google Scholar] [CrossRef]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G., Jr.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef]
- Mix, H.; Widjaja, A.; Jandl, O.; Cornberg, M.; Kaul, A.; Goke, M.; Beil, W.; Kuske, M.; Brabant, G.; Manns, M.P.; et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut 2000, 47, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Guilmeau, S.; Buyse, M.; Bado, A. Gastric leptin: A new manager of gastrointestinal function. Curr. Opin. Pharmacol. 2004, 4, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.M.; Emilsson, V.; Liu, Y.L.; Cawthorne, M.A. Leptin action in intestinal cells. J. Biol. Chem. 1998, 273, 26194–26201. [Google Scholar] [CrossRef]
- Buyse, M.; Berlioz, F.; Guilmeau, S.; Tsocas, A.; Voisin, T.; Peranzi, G.; Merlin, D.; Laburthe, M.; Lewin, M.J.; Roze, C.; et al. Pept1-mediated epithelial transport of dipeptides and cephalexin is enhanced by luminal leptin in the small intestine. J. Clin. Invest. 2001, 108, 1483–1494. [Google Scholar] [CrossRef]
- Attoub, S.; Noe, V.; Pirola, L.; Bruyneel, E.; Chastre, E.; Mareel, M.; Wymann, M.P.; Gespach, C. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho-, and rac-dependent signaling pathways. FASEB J. 2000, 14, 2329–2338. [Google Scholar] [CrossRef]
- Hardwick, J.C.; Van Den Brink, G.R.; Offerhaus, G.J.; Van Deventer, S.J.; Peppelenbosch, M.P. Leptin is a growth factor for colonic epithelial cells. Gastroenterology 2001, 121, 79–90. [Google Scholar] [CrossRef]
- Kiely, J.M.; Noh, J.H.; Pitt, H.A.; Swartz-Basile, D.A. Impaired intestinal cell proliferation and cell death in leptin-deficient obese mice. JPEN J. Parenter. Enteral. Nutr. 2005, 29, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Sukhotnik, I.; Coran, A.G.; Mogilner, J.G.; Shamian, B.; Karry, R.; Lieber, M.; Shaoul, R. Leptin affects intestinal epithelial cell turnover in correlation with leptin receptor expression along the villus-crypt axis after massive small bowel resection in a rat. Pediatr. Res. 2009, 66, 648–653. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rajala, M.W.; Patterson, C.M.; Opp, J.S.; Foltin, S.K.; Young, V.B.; Myers, M.G., Jr. Leptin acts independently of food intake to modulate gut microbial composition in male mice. Endocrinology 2014, 155, 748–757. [Google Scholar] [CrossRef]
- Higurashi, T.; Endo, H.; Uchiyama, T.; Uchiyama, S.; Yamada, E.; Ohkubo, H.; Sakai, E.; Takahashi, H.; Maeda, S.; Wada, K.; et al. Conditional knockout of the leptin receptor in the colonic epithelium revealed the local effects of leptin receptor signaling in the progression of colonic tumors in mice. Carcinogenesis 2014, 35, 2134–2141. [Google Scholar] [CrossRef]
- Guo, X.; Roberts, M.R.; Becker, S.M.; Podd, B.; Zhang, Y.; Chua, S.C., Jr.; Myers, M.G., Jr.; Duggal, P.; Houpt, E.R.; Petri, W.A., Jr. Leptin signaling in intestinal epithelium mediates resistance to enteric infection by entamoeba histolytica. Mucosal. Immunol. 2011, 4, 294–303. [Google Scholar] [CrossRef]
- Sobhani, I.; Bado, A.; Vissuzaine, C.; Buyse, M.; Kermorgant, S.; Laigneau, J.P.; Attoub, S.; Lehy, T.; Henin, D.; Mignon, M.; et al. Leptin secretion and leptin receptor in the human stomach. Gut 2000, 47, 178–183. [Google Scholar] [CrossRef]
- Cinti, S.; Matteis, R.D.; Pico, C.; Ceresi, E.; Obrador, A.; Maffeis, C.; Oliver, J.; Palou, A. Secretory granules of endocrine and chief cells of human stomach mucosa contain leptin. Int. J. Obes. Relat. Metab. Disord 2000, 24, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Azuma, T.; Suto, H.; Ito, Y.; Ohtani, M.; Dojo, M.; Kuriyama, M.; Kato, T. Gastric leptin and helicobacter pylori infection. Gut 2001, 49, 324–329. [Google Scholar] [CrossRef]
- Pai, R.; Lin, C.; Tran, T.; Tarnawski, A. Leptin activates stat and erk2 pathways and induces gastric cancer cell proliferation. BioChem. Biophys Res. Commun. 2005, 331, 984–992. [Google Scholar] [CrossRef]
- Zhao, L.; Shen, Z.X.; Luo, H.S.; Shen, L. Possible involvement of leptin and leptin receptor in developing gastric adenocarcinoma. World J. Gastroenterol. 2005, 11, 7666–7670. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Expression pattern of leptin and leptin receptor (ob-r) in human gastric cancer. World J. Gastroenterol. 2006, 12, 5517–5522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, K.; Zhu, Z.; Chen, S.; Hu, R. Correlation between expression of leptin and clinicopathological features and prognosis in patients with gastric cancer. J. Gastroenterol. Hepatol. 2007, 22, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Choi, H.S.; Yang, S.Y.; Park, H.K.; Lee, Y.Y.; Lee, O.Y.; Yoon, B.C.; Hahm, J.S.; Paik, S.S. The role of leptin in gastric cancer: Clinicopathologic features and molecular mechanisms. BioChem. Biophys Res. Commun. 2014, 446, 822–829. [Google Scholar] [CrossRef]
- Dong, Z.; Fu, S.; Xu, X.; Yang, Y.; Du, L.; Li, W.; Kan, S.; Li, Z.; Zhang, X.; Wang, L.; et al. Leptin-mediated regulation of icam-1 is rho/rock dependent and enhances gastric cancer cell migration. Br. J. Cancer 2014, 110, 1801–1810. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Mayuzumi, H.; Kato, S.; Minokoshi, Y.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Matsuzaki, G.; Yoshimura, A. Enhancement of leptin receptor signaling by socs3 deficiency induces development of gastric tumors in mice. Oncogene 2014, 33, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.C.; Giraud, A.S.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.J.; Malki, S.; Alderman, B.M.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by shp2 and stat-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (ob-r) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Yamachika, T.; Nakanishi, H.; Inada, K.; Tsukamoto, T.; Shimizu, N.; Kobayashi, K.; Fukushima, S.; Tatematsu, M. N-methyl-n-nitrosourea concentration-dependent, rather than total intake-dependent, induction of adenocarcinomas in the glandular stomach of balb/c mice. Jpn. J. Cancer Res. 1998, 89, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Furihata, C.; Ogiu, T.; Tsukamoto, T.; Inada, K.; Hirano, K.; Tatematsu, M. Independent variation in susceptibilities of six different mouse strains to induction of pepsinogen-altered pyloric glands and gastric tumor intestinalization by n-methyl-n-nitrosourea. Cancer Lett. 2002, 179, 121–132. [Google Scholar] [CrossRef]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of helicobacter pylori caga induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef]
- Sakagami, T.; Dixon, M.; O’Rourke, J.; Howlett, R.; Alderuccio, F.; Vella, J.; Shimoyama, T.; Lee, A. Atrophic gastric changes in both helicobacter felis and helicobacter pylori infected mice are host dependent and separate from antral gastritis. Gut 1996, 39, 639–648. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, N.; Choi, Y.J.; Nam, R.H.; Choi, Y.J.; Lee, S.; Choi, D.; Lee, H.S.; Kim, J.W.; Lee, D.H. Effect of n-methyl-n-nitrosourea on helicobacter-induced gastric carcinogenesis in c57bl/6 mice. J. Cancer Prev. 2016, 21, 182–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feng, G.; Zhang, Y.; Yuan, H.; Bai, R.; Zheng, J.; Zhang, J.; Song, M. DNA methylation of trefoil factor 1 (tff1) is associated with the tumorigenesis of gastric carcinoma. Mol. Med. Rep. 2014, 9, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, O.; Chenard, M.P.; Masson, R.; Linares, J.; Dierich, A.; LeMeur, M.; Wendling, C.; Tomasetto, C.; Chambon, P.; Rio, M.C. Gastric mucosa abnormalities and tumorigenesis in mice lacking the ps2 trefoil protein. Science 1996, 274, 259–262. [Google Scholar] [CrossRef]
- Zavros, Y.; Eaton, K.A.; Kang, W.; Rathinavelu, S.; Katukuri, V.; Kao, J.Y.; Samuelson, L.C.; Merchant, J.L. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene 2005, 24, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Judd, L.M.; Andringa, A.; Rubio, C.A.; Spicer, Z.; Shull, G.E.; Miller, M.L. Gastric achlorhydria in h/k-atpase-deficient (atp4a(−/−)) mice causes severe hyperplasia, mucocystic metaplasia and upregulation of growth factors. J. Gastroenterol. Hepatol. 2005, 20, 1266–1278. [Google Scholar] [CrossRef]
- Ray, K.C.; Bell, K.M.; Yan, J.; Gu, G.; Chung, C.H.; Washington, M.K.; Means, A.L. Epithelial tissues have varying degrees of susceptibility to kras(g12d)-initiated tumorigenesis in a mouse model. PLoS ONE 2011, 6, e16786. [Google Scholar] [CrossRef]
- Ota, M.; Horiguchi, M.; Fang, V.; Shibahara, K.; Kadota, K.; Loomis, C.; Cammer, M.; Rifkin, D.B. Genetic suppression of inflammation blocks the tumor-promoting effects of tgf-beta in gastric tissue. Cancer Res. 2014, 74, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; O’Neal, R.; Lee, Y.S.; Lee, Y.C.; Coffey, R.J.; Goldenring, J.R. Gastric tumor development in smad3-deficient mice initiates from forestomach/glandular transition zone along the lesser curvature. Lab. Invest. 2012, 92, 883–895. [Google Scholar] [CrossRef]
- Xu, X.; Brodie, S.G.; Yang, X.; Im, Y.H.; Parks, W.T.; Chen, L.; Zhou, Y.X.; Weinstein, M.; Kim, S.J.; Deng, C.X. Haploid loss of the tumor suppressor smad4/dpc4 initiates gastric polyposis and cancer in mice. Oncogene 2000, 19, 1868–1874. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the wnt signaling and prostaglandin e2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Syder, A.J.; Karam, S.M.; Mills, J.C.; Ippolito, J.E.; Ansari, H.R.; Farook, V.; Gordon, J.I. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 4471–4476. [Google Scholar] [CrossRef]
- Shimada, S.; Mimata, A.; Sekine, M.; Mogushi, K.; Akiyama, Y.; Fukamachi, H.; Jonkers, J.; Tanaka, H.; Eishi, Y.; Yuasa, Y. Synergistic tumour suppressor activity of e-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut 2012, 61, 344–353. [Google Scholar] [CrossRef]
- Lee, M.P.; Ravenel, J.D.; Hu, R.J.; Lustig, L.R.; Tomaselli, G.; Berger, R.D.; Brandenburg, S.A.; Litzi, T.J.; Bunton, T.E.; Limb, C.; et al. Targeted disruption of the kvlqt1 gene causes deafness and gastric hyperplasia in mice. J. Clin. Invest. 2000, 106, 1447–1455. [Google Scholar] [CrossRef]
- Ito, K.; Chuang, L.S.; Ito, T.; Chang, T.L.; Fukamachi, H.; Salto-Tellez, M.; Ito, Y. Loss of runx3 is a key event in inducing precancerous state of the stomach. Gastroenterology 2011, 140, 1536–1546.e8. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Okamoto, S.; Takagi, K.; Saito, K.; Arita, S.; Tang, L.; Hori, T.; Kataoka, H.; Matsumoto, S.; Minokoshi, Y. Leptin receptor signaling is required for high-fat diet-induced atrophic gastritis in mice. Nutr. Metab. 2016, 13. [Google Scholar] [CrossRef]
- Arita, S.; Kinoshita, Y.; Ushida, K.; Enomoto, A.; Inagaki-Ohara, K. High-fat diet feeding promotes stemness and precancerous changes in murine gastric mucosa mediated by leptin receptor signaling pathway. Arch. BioChem. Biophys 2016, 610, 16–24. [Google Scholar] [CrossRef]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu. Rev. BioChem. 1977, 46, 897–930. [Google Scholar] [CrossRef]
- Las, G.; Serada, S.B.; Wikstrom, J.D.; Twig, G.; Shirihai, O.S. Fatty acids suppress autophagic turnover in beta-cells. J. Biol. Chem. 2011, 286, 42534–42544. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Matsusaka, T.; et al. High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J. Am. Soc. Nephrol. 2017, 28, 1534–1551. [Google Scholar] [CrossRef]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front. Endocrinol. (Lausanne) 2018, 9. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting hif-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Velloso, L.A. Hypoxia inducible factor as a central regulator of metabolism—Implications for the development of obesity. Front. NeuroSci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, S.; Yazdanparast, R. Activation of stat3/hif-1alpha/hes-1 axis promotes trastuzumab resistance in her2-overexpressing breast cancer cells via down-regulation of pten. Biochim. Biophys Acta Gen. Subj. 2017, 1861, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Ueo, T.; Fukuda, A.; Kawada, K.; Sakai, Y.; Miyoshi, H.; Taketo, M.M.; Chiba, T.; Seno, H. Distinct roles of hes1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Res. 2017, 77, 3442–3454. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; deGraffenried, L.A.; Hursting, S.D. Leptin signaling mediates obesity-associated csc enrichment and emt in preclinical tnbc models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef]
- Haque, I.; Ghosh, A.; Acup, S.; Banerjee, S.; Dhar, K.; Ray, A.; Sarkar, S.; Kambhampati, S.; Banerjee, S.K. Leptin-induced er-alpha-positive breast cancer cell viability and migration is mediated by suppressing ccn5-signaling via activating jak/akt/stat-pathway. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Xu, M.; Cao, F.L.; Li, N.; Gao, X.; Su, X.; Jiang, X. Leptin induces epithelial-to-mesenchymal transition via activation of the erk signaling pathway in lung cancer cells. Oncol. Lett. 2018, 16, 4782–4788. [Google Scholar] [CrossRef]
- Kimelman, D.; Xu, W. Beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, R.L.; Xu, A.M. Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res. 2015, 7, 2141–2158. [Google Scholar]
- Rassouli, F.B.; Matin, M.M.; Saeinasab, M. Cancer stem cells in human digestive tract malignancies. Tumour Biol. 2016, 37, 7–21. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Kunz, P.L.; Gubens, M.; Fisher, G.A.; Ford, J.M.; Lichtensztajn, D.Y.; Clarke, C.A. Long-term survivors of gastric cancer: A california population-based study. J. Clin. Oncol. 2012, 30, 3507–3515. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of her2-positive advanced gastric or gastro-oesophageal junction cancer (toga): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. Her2 testing in gastric cancer: An update. World J. Gastroenterol. 2016, 22, 4619–4625. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (regard): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Bain, G.H.; Collie-Duguid, E.; Murray, G.I.; Gilbert, F.J.; Denison, A.; McKiddie, F.; Ahearn, T.; Fleming, I.; Leeds, J.; Phull, P.; et al. Tumour expression of leptin is associated with chemotherapy resistance and therapy-independent prognosis in gastro-oesophageal adenocarcinomas. Br. J. Cancer 2014, 110, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Blank, S.; Deck, C.; Dreikhausen, L.; Weichert, W.; Giese, N.; Falk, C.; Schmidt, T.; Ott, K. Angiogenic and growth factors in gastric cancer. J. Surg. Res. 2015, 194, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin-notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget 2018, 9, 18239–18253. [Google Scholar] [CrossRef] [PubMed]
- Raut, P.K.; Kim, S.H.; Choi, D.Y.; Jeong, G.S.; Park, P.H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. BioChem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Alti, D.; Sambamurthy, C.; Kalangi, S.K. Emergence of leptin in infection and immunity: Scope and challenges in vaccines formulation. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Model | Incidence (%) | Periods or Age Onset | Phenotype | Causative Mechanism | References No. |
|---|---|---|---|---|---|
| Chemical carcinogen/Bacterial infection induced | |||||
| MNU | (1) 60% (2) 19% | > 50 weeks | Adenocarcinoma | An alkylating reagent in experimental gastric carcinogenesis | (1) [65] (2) [66] |
| H. felis | 80% | < 6months | Severe gastritis | More susceptible Helicobacter spp for C57BL/6 murine model | [68] |
| MNU + H. felis | 40% | 9 months | Adenoma | [69] | |
| Gene-targeting | |||||
| pS2 (TFF1)−/− | 30% | 5 months | Dysplasia | Lacking normal gastric mucus | [71] |
| Gan (K19-Wnt1/C2mETg) | 100% | < 10 months | Carcinoma | Excess of COX-2 and microsomal prostaglandin E synthase-1 | [79] |
| INS-GAS | 85% | > 20 months | Intramucosal carcinoma | Hyperexpression of gastrin | [80] |
| INS-GAS + H. felis | < 8 months | ||||
| GAS−/− | 60% | 1 year | Displasia | Lacking gastrin | [72] |
| Atp4a−/− | 100% | 1 year | Incomplete intestinal metaplasia | Lacking H+K+ ATPase | [73] |
| Atp4b-SV40 | 60% | 1 year | Carcinoma, invasion (Lymphatic–vascular), metastasis (liver) | Expression of SV40 in parietal cells | [81] |
| Atp4b- (CDH1xTrp53)−/− | 100% | 1 year | Metastasized to lymph nodes | Lacking E-cadherin and p53 in parietal cells | [82] |
| ATP4b-hIL-1b | 30% | 1 year | Dysplasia, Adenocarcinoma | MDSCs recruitment via IL-1RI/NF-κB pathway | [78] |
| Kvlqt1−/− | 100% | 3 months | Hyperplasia in gastric neck cells | Lacking potassium channel | [83] |
| K-ras G12D (systemic) | 100% | < 18 days | Metaplasia | Hyperactivation of MAPK by K-ras mutation | [74] |
| Tgfβ1-C33S | 40% | 4-5 months | Well-differentiated adenocarcinomas | Unable forming latent TGF-β binding protein-1 | [75] |
| Smad3−/− | 100% | 10 months | Tumors, invasive neoplasia | Excess of cytosolic E-cadherin | [76] |
| Smad4−/− | 100% | > 1 year | Invasive carcinoma | Increased cyclin1 and upregulation of TGF-β1 | [77] |
| RUNX3−/− | 70% | > 1 year | Hyperplasia, Chief cells loss and increased cdx2 | Enhanced Wnt-β catenin signaling by RUNX3 loss | [84] |
| gp130757F | 100% | 3 months | Adenoma | Abrogating SHP2-Ras-ERK signaling | [63] |
| T3b-SOCS3−/− | 100% | 2 months | Carcinoma | Augment of leptin expression and ObR-STAT3 signaling by gastrointestinal cell- specific SOCS3 loss | [62] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inagaki-Ohara, K. Gastric Leptin and Tumorigenesis: Beyond Obesity. Int. J. Mol. Sci. 2019, 20, 2622. https://doi.org/10.3390/ijms20112622
Inagaki-Ohara K. Gastric Leptin and Tumorigenesis: Beyond Obesity. International Journal of Molecular Sciences. 2019; 20(11):2622. https://doi.org/10.3390/ijms20112622
Chicago/Turabian StyleInagaki-Ohara, Kyoko. 2019. "Gastric Leptin and Tumorigenesis: Beyond Obesity" International Journal of Molecular Sciences 20, no. 11: 2622. https://doi.org/10.3390/ijms20112622
APA StyleInagaki-Ohara, K. (2019). Gastric Leptin and Tumorigenesis: Beyond Obesity. International Journal of Molecular Sciences, 20(11), 2622. https://doi.org/10.3390/ijms20112622