Gastric Cancer Extracellular Vesicles Tune the Migration and Invasion of Epithelial and Mesenchymal Cells in a Histotype-Dependent Manner

 , ,
, ,
Abstract
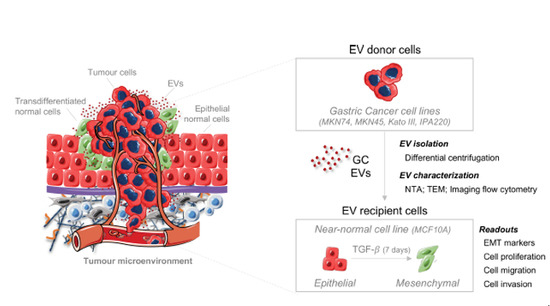
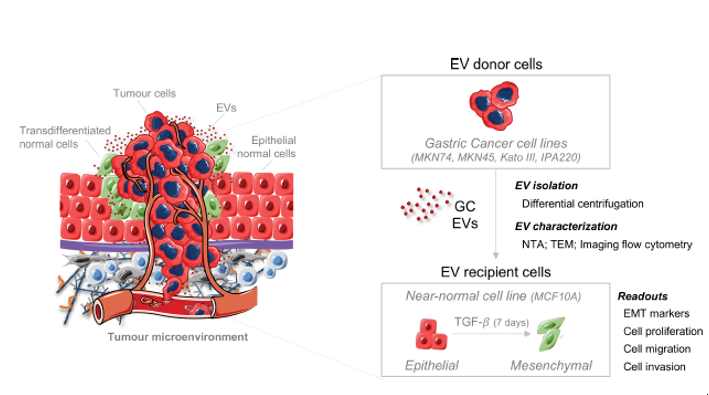{kind=link}
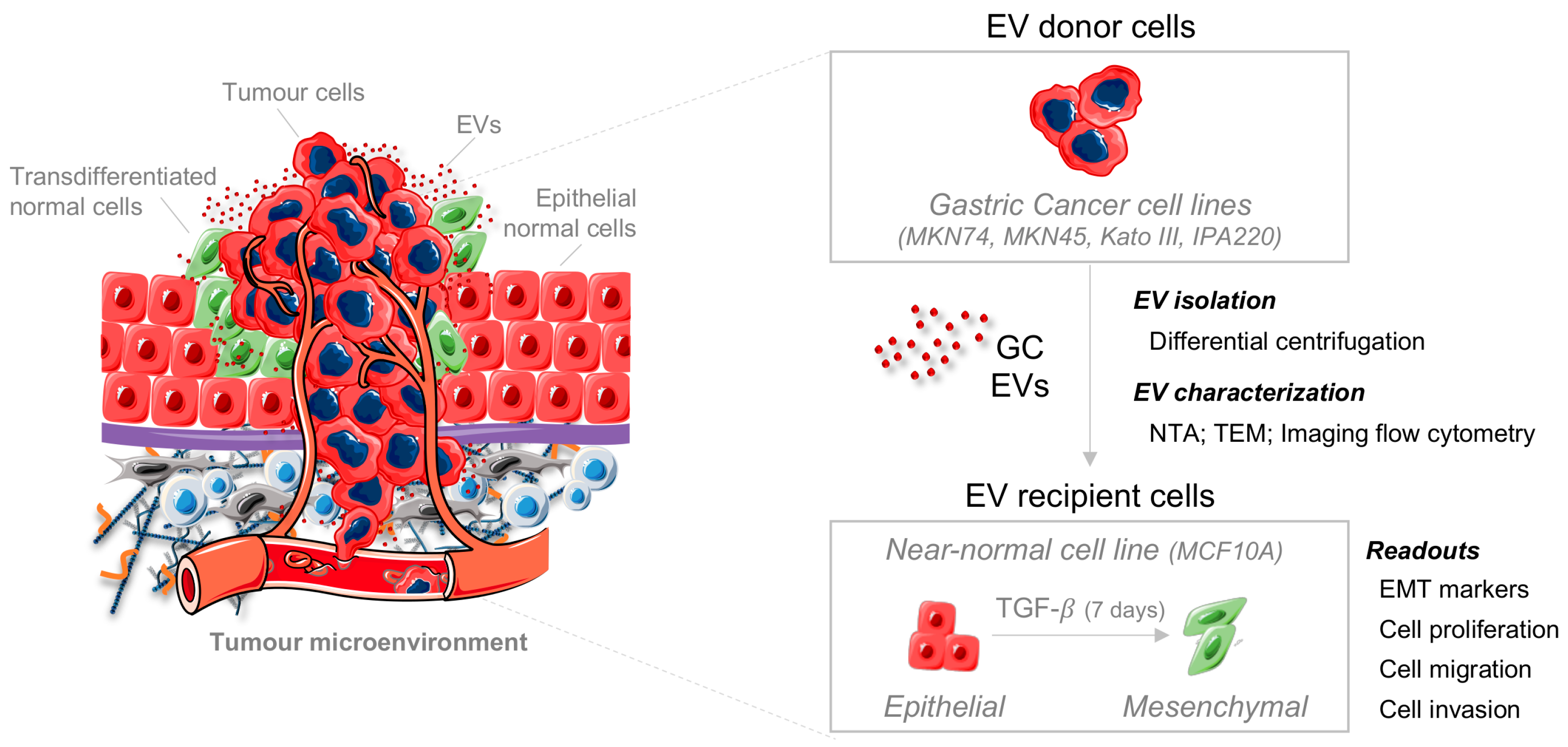{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TGFβ1 Induces EMT in a Non-Tumorigenic Epithelial Cell Line
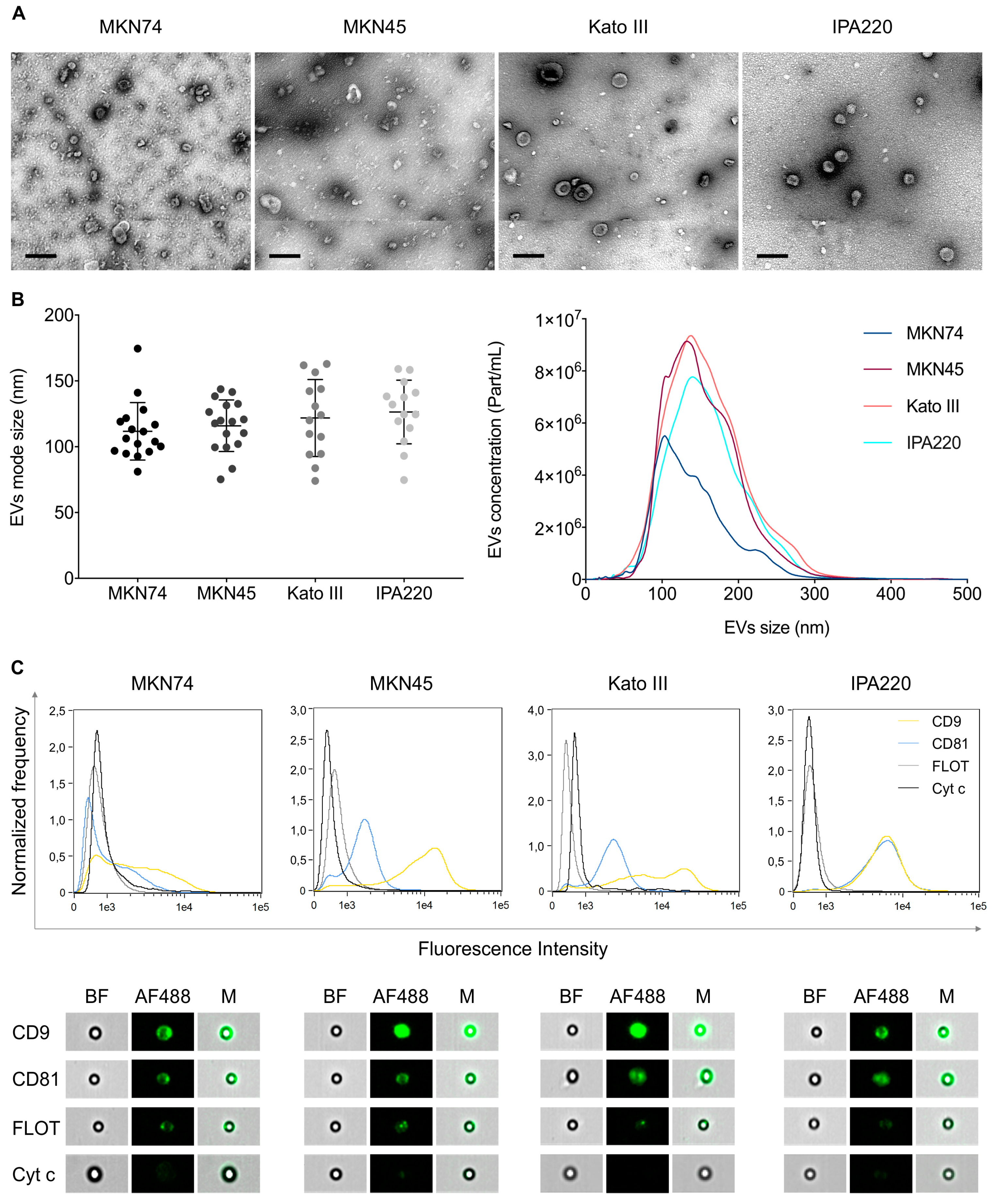
2.2. Distinct GC Cell Lines Secrete EVs with Similar Physical and Biochemical Properties
2.3. GC-EVs Do Not Modulate E-Cadherin Expression at the Cell Membrane
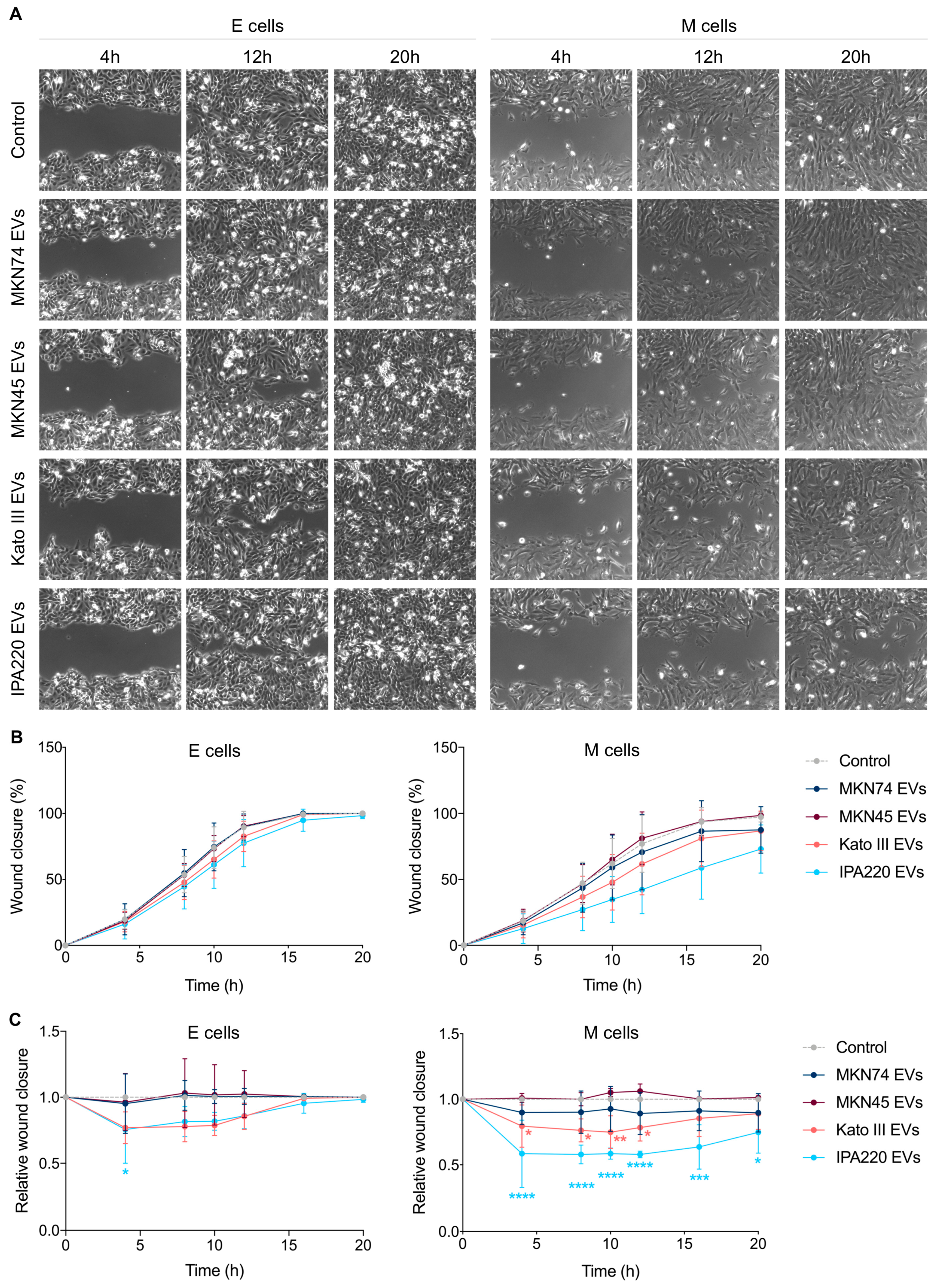
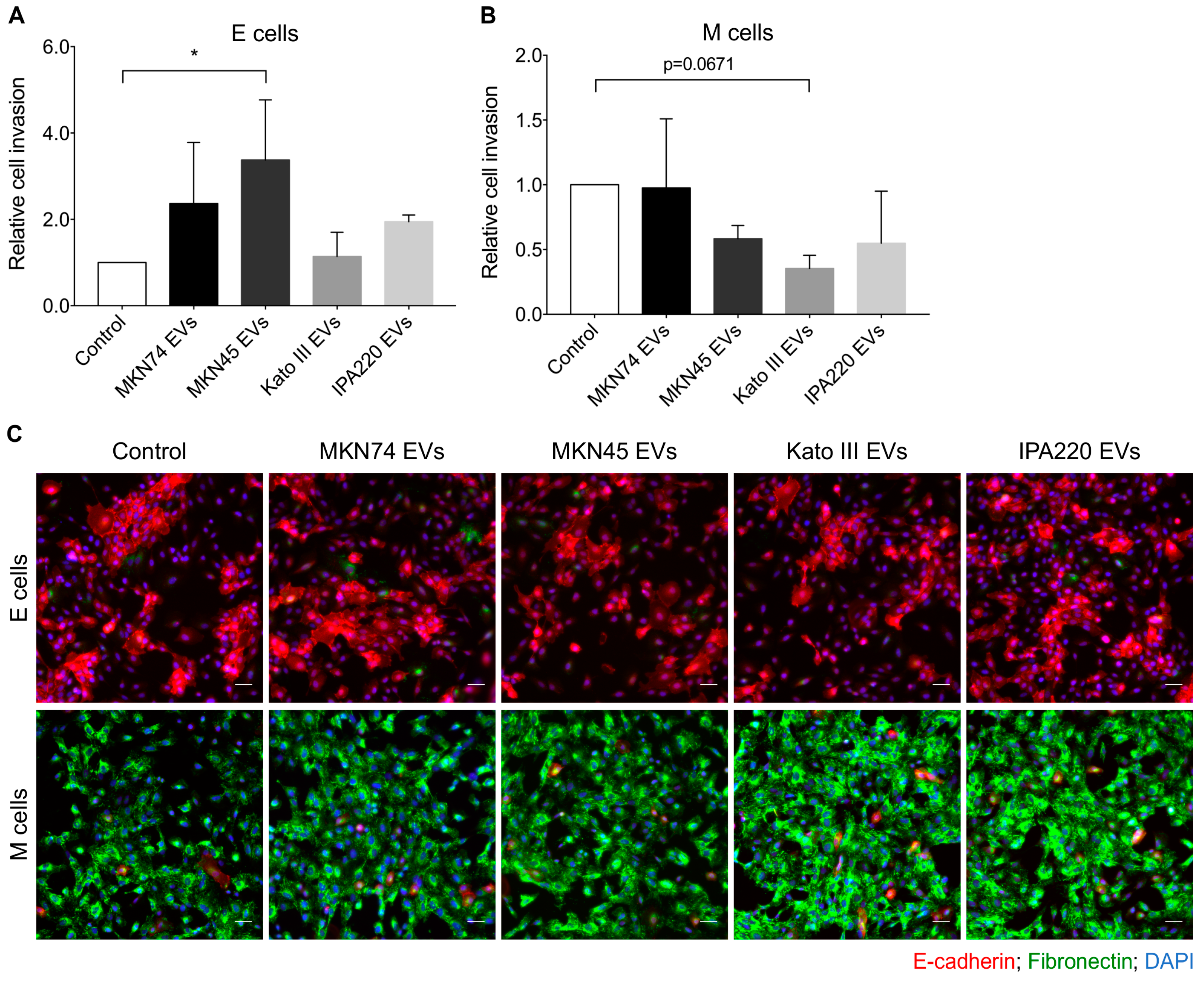
2.4. GC-EVs Impair Migration and Invasion of Epithelial and Mesenchymal Cells
3. Discussion
4. Materials and Methods
4.1. Establishment and Characterization of Epithelial-to-Mesenchymal Transition EMT Recipient Model
4.1.1. Treatment of MCF10A Epithelial Cell Line with TGFβ1
4.1.2. Co- Immunofluorescence of E-Cadherin and Fibronectin
4.1.3. RNA Expression Quantification of Epithelial and Mesenchymal Markers
4.2. Characterization of GC-EVs
4.2.1. GC Cell Culture and Viability
4.2.2. EV Isolation by Differential Centrifugation
4.2.3. EV Morphology by Transmission Electron Microscopy (TEM)
4.2.4. EV Characterization by Nanoparticle Tracking Analysis (NTA)
4.2.5. EV Characterization by Imaging Flow Cytometry
4.3. Functional Assays with Recipient Cells Treated with GC-EVs
4.3.1. Treatment of Epithelial and Mesenchymal Cells with GC-EVs
4.3.2. Flow Cytometry Analysis of E-Cadherin
4.3.3. Wound-Healing Migration Assay
4.3.4. Proliferation Assay
4.3.5. Matrigel Invasion Assay
4.3.6. Co-Immunofluorescence of E-Cadherin and Fibronectin
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EVs | Extracellular vesicles |
| EMT | Epithelial-to-mesenchymal transition |
| GC | Gastric cancer |
| GC-EVs | Gastric cancer-derived extracellular vesicles |
| MSCs | Mesenchymal stem cells |
| CAFs | Cancer-associated fibroblasts |
| MMT | Mesothelial-to-mesenchymal transition |
| TEM | Transmission electron microscopy |
| NTA | Nanoparticle tracking analysis |
| E | Epithelial cells |
| M | Mesenchymal cells |
References
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-based cell-cell communication in the tumor microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, J.N.; Demory Beckler, M.; Gephart, J.D.; Franklin, J.L.; Bogatcheva, G.; Kremers, G.J.; Piston, D.W.; Ayers, G.D.; McConnell, R.E.; Tyska, M.J.; et al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 2011, 21, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schafer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, R.H.; Foreman, K.E.; Gupta, G.N. The role of cancer-derived exosomes in tumorigenicity & epithelial-to-mesenchymal transition. Cancers 2017, 9, 105. [Google Scholar]
- Franzen, C.A.; Blackwell, R.H.; Todorovic, V.; Greco, K.A.; Foreman, K.E.; Flanigan, R.C.; Kuo, P.C.; Gupta, G.N. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis 2015, 4, e163. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef]
- Rahman, M.A.; Barger, J.F.; Lovat, F.; Gao, M.; Otterson, G.A.; Nana-Sinkam, P. Lung cancer exosomes as drivers of epithelial mesenchymal transition. Oncotarget 2016, 7, 54852–54866. [Google Scholar] [CrossRef]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal micrornas that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wang, J.; Li, M.; Cao, C.; Tan, J.; Ma, D.; Gao, Q. TGFbeta1 in fibroblasts-derived exosomes promotes epithelial-mesenchymal transition of ovarian cancer cells. Oncotarget 2017, 8, 96035–96047. [Google Scholar] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-pcp signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef]
- Yan, Y.; Fu, G.; Ye, Y.; Ming, L. Exosomes participate in the carcinogenesis and the malignant behavior of gastric cancer. Scand. J. Gastroenterol. 2017, 52, 499–504. [Google Scholar] [CrossRef]
- Wang, J.; Guan, X.; Zhang, Y.; Ge, S.; Zhang, L.; Li, H.; Wang, X.; Liu, R.; Ning, T.; Deng, T.; et al. Exosomal miR-27a derived from gastric cancer cells regulates the transformation of fibroblasts into cancer-associated fibroblasts. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 869–883. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, H.; Wang, C.; Song, X. Exosomes released by gastric cancer cells induce transition of pericytes into cancer-associated fibroblasts. Med Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 2350–2359. [Google Scholar] [CrossRef]
- Gu, J.; Qian, H.; Shen, L.; Zhang, X.; Zhu, W.; Huang, L.; Yan, Y.; Mao, F.; Zhao, C.; Shi, Y.; et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-beta/smad pathway. PloS ONE 2012, 7, e52465. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xue, C.; Li, X.; Ba, L.; Gu, J.; Sun, Z.; Han, Q.; Zhao, R.C. Effects of gastric cancer cell-derived exosomes on the immune regulation of mesenchymal stem cells by the NF-κB signaling pathway. Stem Cells Dev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.; Zhang, B.; Shi, H.; Yuan, X.; Sun, Y.; Pan, Z.; Qian, H.; Xu, W. Exosomes derived from gastric cancer cells activate nf-kappab pathway in macrophages to promote cancer progression. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 12169–12180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce n2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef]
- Miki, Y.; Yashiro, M.; Okuno, T.; Kitayama, K.; Masuda, G.; Hirakawa, K.; Ohira, M. Cd9-positive exosomes from cancer-associated fibroblasts stimulate the migration ability of scirrhous-type gastric cancer cells. Br. J. Cancer 2018, 118, 867–877. [Google Scholar] [CrossRef]
- Gu, H.; Ji, R.; Zhang, X.; Wang, M.; Zhu, W.; Qian, H.; Chen, Y.; Jiang, P.; Xu, W. Exosomes derived from human mesenchymal stem cells promote gastric cancer cell growth and migration via the activation of the akt pathway. Mol. Med. Rep. 2016, 14, 3452–3458. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through met. Nature Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered egfr regulates liver microenvironment to promote gastric cancer liver metastasis. Nature Commun. 2017, 8, 15016. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Qu, J.; Zhang, Y.; Che, X.; Cheng, Y.; Fan, Y.; Zhang, S.; Na, D.; Liu, Y.; Qu, X. Gastric cancer-derived exosomes promote peritoneal metastasis by destroying the mesothelial barrier. FEBS Lett. 2017, 591, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, B.; Li, Q.; Wei, S.; He, Z.; Huang, X.; Wang, L.; Xia, Y.; Xu, Z.; Li, Z.; et al. Exosomal mir-21-5p derived from gastric cancer promotes peritoneal metastasis via mesothelial-to-mesenchymal transition. Cell Death Dis. 2018, 9, 854. [Google Scholar] [CrossRef]
- Motoyama, T.; Hojo, H.; Watanabe, H. Comparison of seven cell lines derived from human gastric carcinomas. Acta Pathol. Jpn. 1986, 36, 65–83. [Google Scholar] [CrossRef]
- Gartner, F.; David, L.; Seruca, R.; Machado, J.C.; Sobrinho-Simoes, M. Establishment and characterization of two cell lines derived from human diffuse gastric carcinomas xenografted in nude mice. Virchows Archiv. Int. J. Pathol. 1996, 428, 91–98. [Google Scholar] [CrossRef]
- Pinho, S.S.; Oliveira, P.; Cabral, J.; Carvalho, S.; Huntsman, D.; Gartner, F.; Seruca, R.; Reis, C.A.; Oliveira, C. Loss and recovery of mgat3 and gnt-iii mediated e-cadherin n-glycosylation is a mechanism involved in epithelial-mesenchymal-epithelial transitions. PloS ONE 2012, 7, e33191. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 3–22. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Ribeiro, A.S.; Sousa, B.; Carreto, L.; Mendes, N.; Nobre, A.R.; Ricardo, S.; Albergaria, A.; Cameselle-Teijeiro, J.F.; Gerhard, R.; Soderberg, O.; et al. P-cadherin functional role is dependent on e-cadherin cellular context: A proof of concept using the breast cancer model. J. Pathol. 2013, 229, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, C.X.; Fang, E.H.; Wang, G.B.; Tong, Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J. Gastroenterol. 2014, 20, 5403–5410. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary diffuse gastric cancer syndrome: Cdh1 mutations and beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Duarte, I.; Llanos, O. Patterns of metastases in intestinal and diffuse types of carcinoma of the stomach. Human Pathol. 1981, 12, 237–242. [Google Scholar] [CrossRef]
- Riihimaki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Metastatic spread in patients with gastric cancer. Oncotarget 2016, 7, 52307–52316. [Google Scholar] [CrossRef]
- Li, W.; Ng, J.M.; Wong, C.C.; Ng, E.K.W.; Yu, J. Molecular alterations of cancer cell and tumour microenvironment in metastatic gastric cancer. Oncogene 2018, 37, 4903–4920. [Google Scholar] [CrossRef]
- Arita, T.; Ichikawa, D.; Konishi, H.; Komatsu, S.; Shiozaki, A.; Ogino, S.; Fujita, Y.; Hiramoto, H.; Hamada, J.; Shoda, K.; et al. Tumor exosome-mediated promotion of adhesion to mesothelial cells in gastric cancer cells. Oncotarget 2016, 7, 56855–56863. [Google Scholar] [CrossRef] [PubMed]
- Sakha, S.; Muramatsu, T.; Ueda, K.; Inazawa, J. Exosomal microrna mir-1246 induces cell motility and invasion through the regulation of dennd2d in oral squamous cell carcinoma. Scientific Rep. 2016, 6, 38750. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A.; Patel, S.H.; Gucek, M.; Hendrix, A.; Westbroek, W.; Taraska, J.W. Exosomes released from breast cancer carcinomas stimulate cell movement. PloS ONE 2015, 10, e0117495. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Carvalho, J.; Oliveira, P.; Voglstaetter, M.; Schvartz, D.; Thomsen, A.R.; Walter, N.; Khanduri, R.; Sanchez, J.C.; Keller, A.; et al. 3d cellular architecture affects microrna and protein cargo of extracellular vesicles. Adv. Sci. 2019, 6, 1800948. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; van Grieken, N.C.; Pereira, P.M.; Sousa, S.; Tijssen, M.; Buffart, T.E.; Diosdado, B.; Grabsch, H.; Santos, M.A.; Meijer, G.; et al. Lack of microrna-101 causes e-cadherin functional deregulation through ezh2 up-regulation in intestinal gastric cancer. J. Pathol. 2012, 228, 31–44. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, S.; Teles, S.P.; Azevedo, M.; Oliveira, P.; Carvalho, J.; Oliveira, C. Gastric Cancer Extracellular Vesicles Tune the Migration and Invasion of Epithelial and Mesenchymal Cells in a Histotype-Dependent Manner. Int. J. Mol. Sci. 2019, 20, 2608. https://doi.org/10.3390/ijms20112608
Rocha S, Teles SP, Azevedo M, Oliveira P, Carvalho J, Oliveira C. Gastric Cancer Extracellular Vesicles Tune the Migration and Invasion of Epithelial and Mesenchymal Cells in a Histotype-Dependent Manner. International Journal of Molecular Sciences. 2019; 20(11):2608. https://doi.org/10.3390/ijms20112608
Chicago/Turabian StyleRocha, Sara, Sara Pinto Teles, Mafalda Azevedo, Patrícia Oliveira, Joana Carvalho, and Carla Oliveira. 2019. "Gastric Cancer Extracellular Vesicles Tune the Migration and Invasion of Epithelial and Mesenchymal Cells in a Histotype-Dependent Manner" International Journal of Molecular Sciences 20, no. 11: 2608. https://doi.org/10.3390/ijms20112608
APA StyleRocha, S., Teles, S. P., Azevedo, M., Oliveira, P., Carvalho, J., & Oliveira, C. (2019). Gastric Cancer Extracellular Vesicles Tune the Migration and Invasion of Epithelial and Mesenchymal Cells in a Histotype-Dependent Manner. International Journal of Molecular Sciences, 20(11), 2608. https://doi.org/10.3390/ijms20112608