Endocannabinoid System in Hepatic Glucose Metabolism, Fatty Liver Disease, and Cirrhosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Expression of Cannabinoid Receptors CB1 and CB2
2.1. Diurnal Expression of Hepatic Cannabinoid Receptors CB1 and CB2
2.2. Cannabinoid Receptor CB1- and CB2-Function and Its Metabolic Consequences in the Liver
2.3. Cannabinoid Receptor CB1- and CB2-Agonism/Antagonism in Metabolic Disorders of the Liver
3. Other Hepatic Cannabinoid Receptors in Metabolism and Metabolic Disorders of the Liver
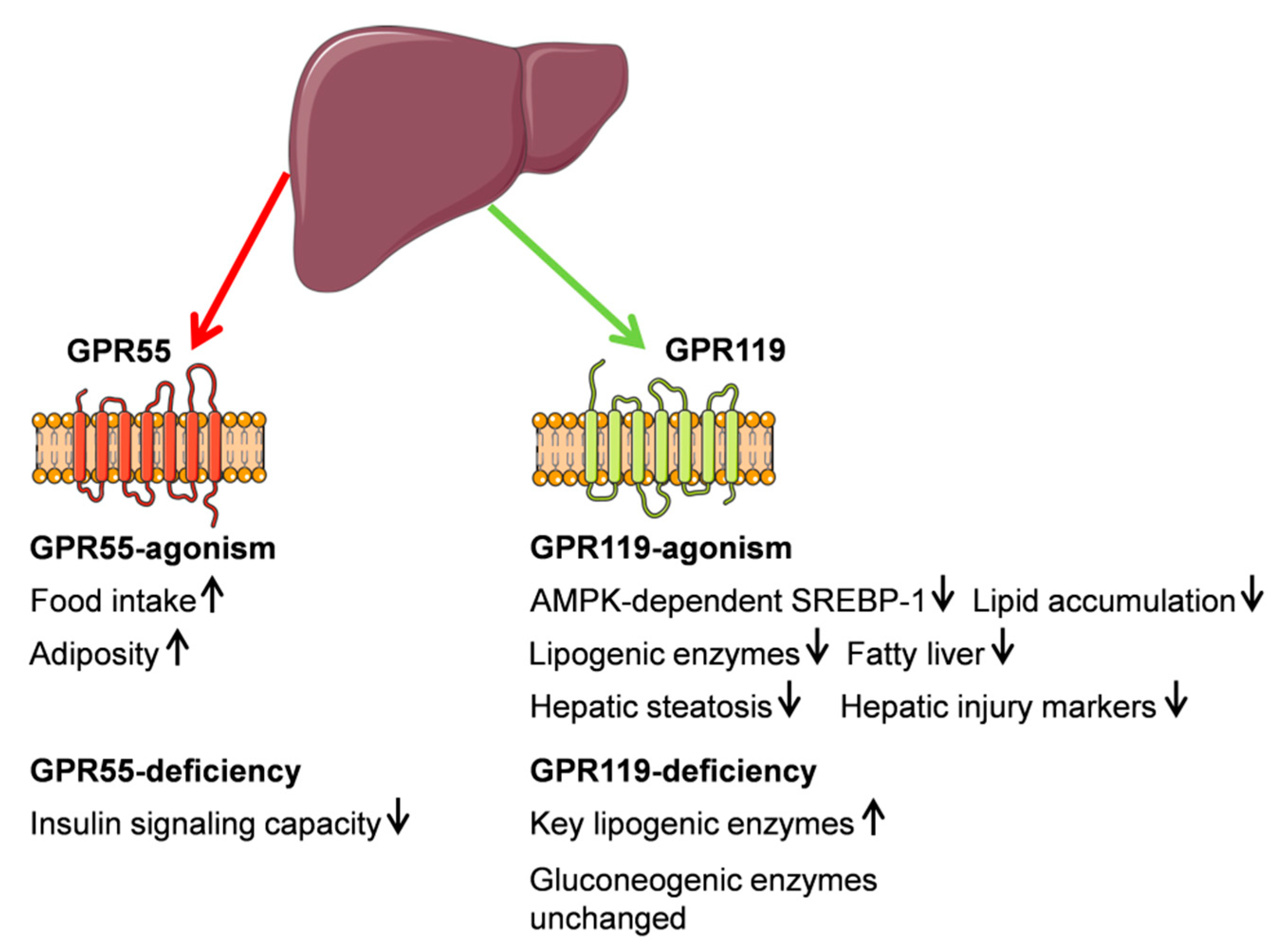
3.1. G Protein-Coupled Receptor 55 (GPR55)
3.2. G Protein-Coupled Receptor (GPR119)
4. Peroxisome Proliferator-Activated Receptors (PPARs)
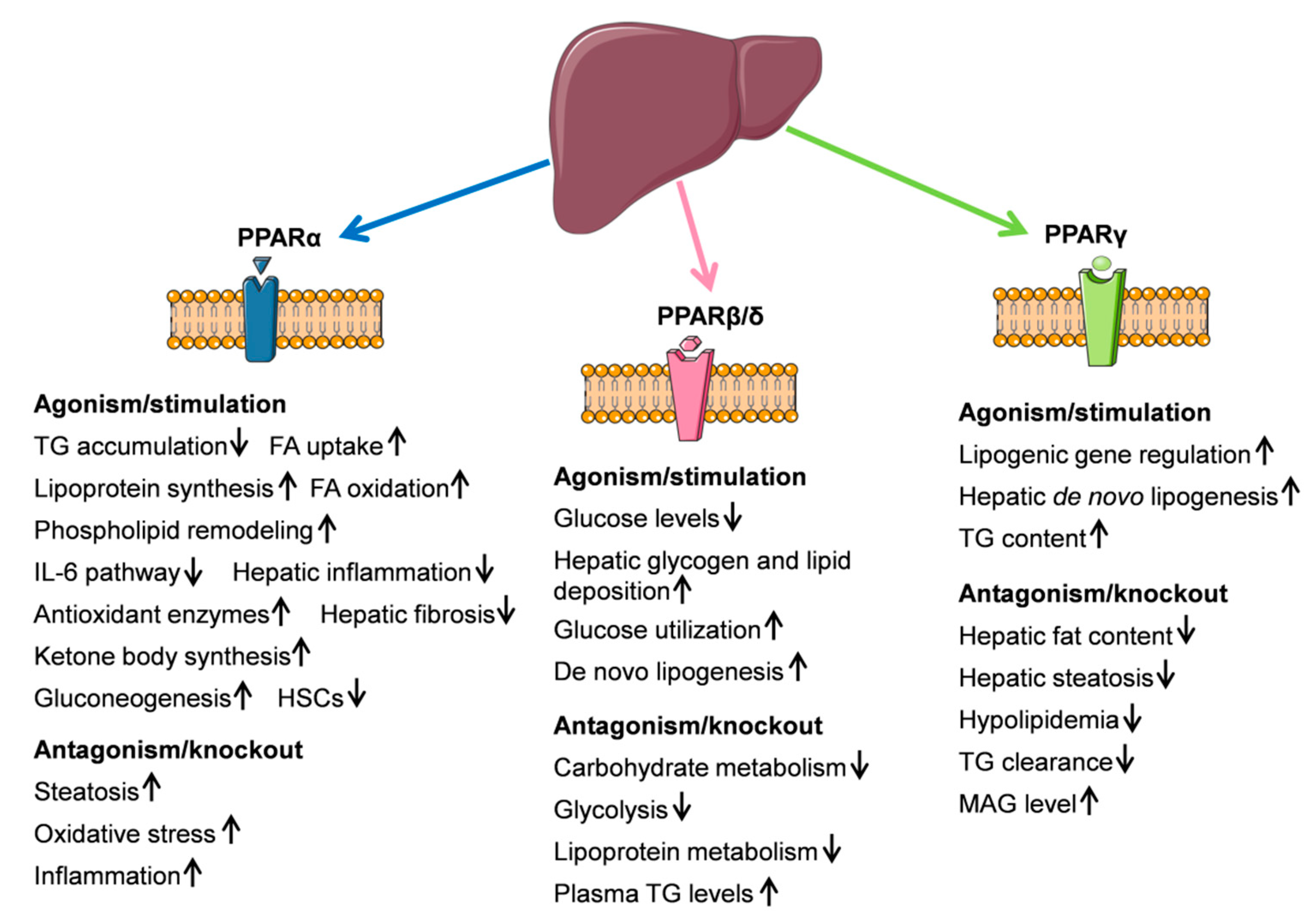
4.1. PPARα
4.2. PPARβ/δ
4.3. PPARγ
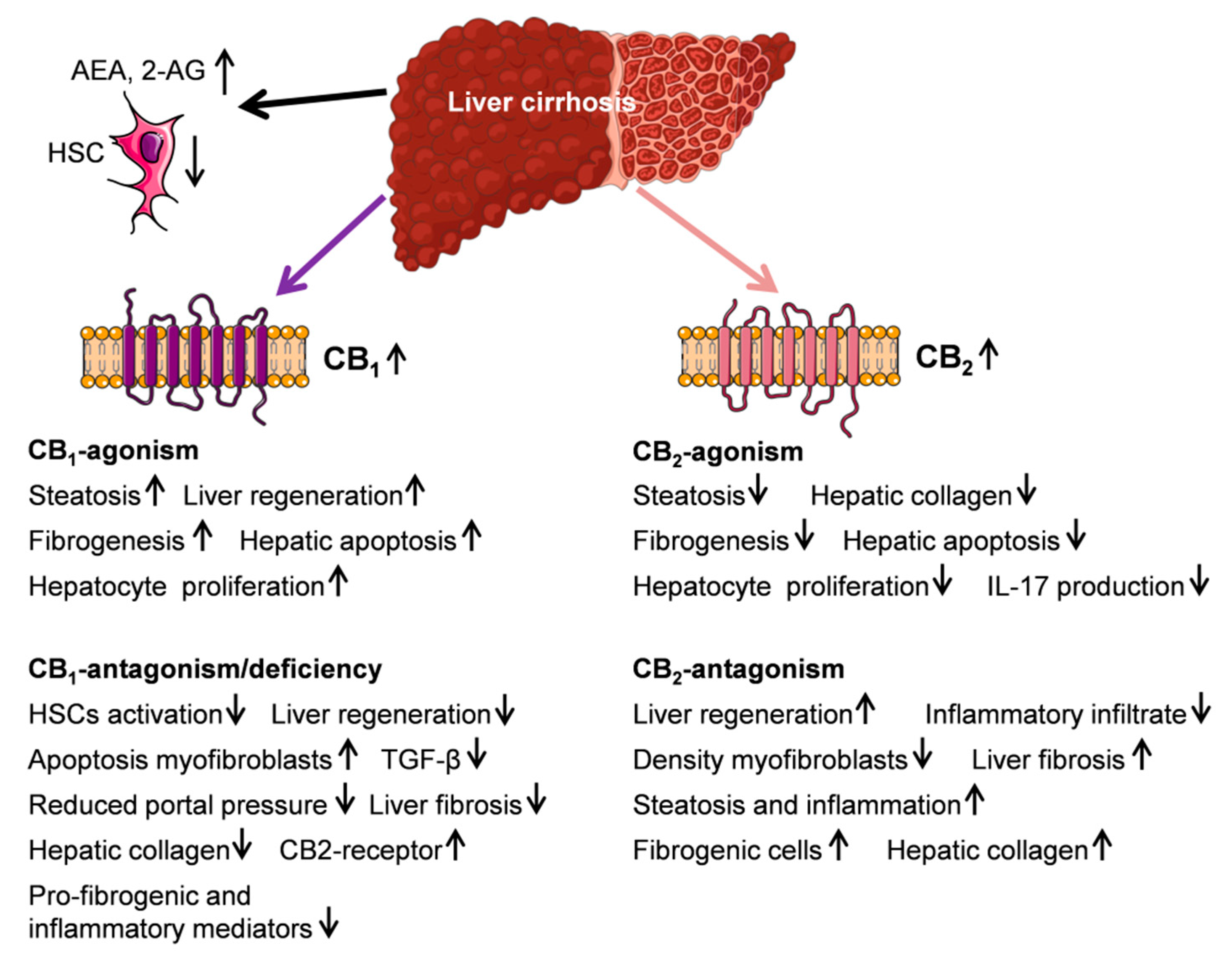
5. Endocannabinoid System and Liver Cirrhosis
5.1. Endocannabinoids AEA, 2-AG, and PEA
5.2. CB1- and CB2-Receptor
5.3. Endocannabinoids and Hemodynamics in Cirrhosis
6. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simon, V.; Cota, D. MECHANISMS IN ENDOCRINOLOGY: Endocannabinoids and metabolism: Past, present and future. Eur. J. Endocrinol. 2017, 176, R309–R324. [Google Scholar] [CrossRef] [PubMed]
- de Petrocellis, L.; Cascio, M.G.; Di Marzo, V. The endocannabinoid system: A general view and latest additions. Br. J. Pharmacol. 2004, 141, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Breivogel, C.S.; Childers, S.R.; Deadwyler, S.A.; Hampson, R.E.; Porrino, L.J. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology 2004, 47, 345–358. [Google Scholar] [CrossRef]
- Di Marzo, V. CB(1) receptor antagonism: Biological basis for metabolic effects. Drug Discov. Today 2008, 13, 1026–1041. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB₁ and CB₂. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef]
- Bouaboula, M.; Hilairet, S.; Marchand, J.; Fajas, L.; Le Fur, G.; Casellas, P. Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 2005, 517, 174–181. [Google Scholar] [CrossRef]
- Astarita, G.; Ahmed, F.; Piomelli, D. Identification of biosynthetic precursors for the endocannabinoid anandamide in the rat brain. J. Lipid Res. 2008, 49, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Godlewski, G.; Kunos, G. Endocannabinoid regulation of β-cell functions: Implications for glycaemic control and diabetes. DiabetesObes. Metab. 2016, 18, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Buckley, N.E.; McCoy, K.L.; Mezey, E.; Bonner, T.; Zimmer, A.; Felder, C.C.; Glass, M. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur. J. Pharmacol. 2000, 396, 141–149. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An Update on Non-CB1, Non-CB2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Dalton, G.D.; Bass, C.E.; van Horn, C.G.; Howlett, A.C. Signal transduction via cannabinoid receptors. CNS Neurol. Disord. Drug Targets 2009, 8, 422–431. [Google Scholar] [CrossRef]
- van Sickle, M.D. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Skaper, S.D.; Buriani, A.; Dal Toso, R.; Petrelli, L.; Romanello, S.; Facci, L.; Leon, A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 3984–3989. [Google Scholar] [CrossRef]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: An immunohistochemical study. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef]
- Massa, F.; Storr, M.; Lutz, B. The endocannabinoid system in the physiology and pathophysiology of the gastrointestinal tract. J. Mol. Med. 2005, 83, 944–954. [Google Scholar] [CrossRef]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bákai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef]
- Cota, D.; Genghini, S.; Pasquali, R.; Pagotto, U. Antagonizing the cannabinoid receptor type 1: A dual way to fight obesity. J. Endocrinol. Investig. 2003, 26, 1041–1044. [Google Scholar] [CrossRef]
- Tharp, W.G.; Lee, Y.-H.; Maple, R.L.; Pratley, R.E. The cannabinoid CB1 receptor is expressed in pancreatic delta-cells. Biochem. Biophys. Res. Commun. 2008, 372, 595–600. [Google Scholar] [CrossRef]
- O’Keefe, L.; Simcocks, A.C.; Hryciw, D.H.; Mathai, M.L.; McAinch, A.J. The cannabinoid receptor 1 and its role in influencing peripheral metabolism. Diabetes Obes. Metab. 2014, 16, 294–304. [Google Scholar] [CrossRef]
- Brown, S.M.; Wager-Miller, J.; Mackie, K. Cloning and molecular characterization of the rat CB2 cannabinoid receptor. Biochim. Biophys. Acta 2002, 1576, 255–264. [Google Scholar] [CrossRef]
- Julien, B.; Grenard, P.; Teixeira-Clerc, F.; van Nhieu, J.T.; Li, L.; Karsak, M.; Zimmer, A.; Mallat, A.; Lotersztajn, S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Zamora-Valdes, D.; Pichardo-Bahena, R.; Barredo-Prieto, B.; Ponciano-Rodriguez, G.; Bermejo-Martínez, L.; Chavez-Tapia, N.C.; Baptista-González, H.A.; Uribe, M. Endocannabinoid receptor CB2 in nonalcoholic fatty liver disease. Liver Int. 2007, 27, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Teixeira-Clerc, F.; Julien, B.; Grenard, P.; van Tran Nhieu, J.; Deveaux, V.; Li, L.; Serriere-Lanneau, V.; Ledent, C.; Mallat, A.; Lotersztajn, S. CB1 cannabinoid receptor antagonism: A new strategy for the treatment of liver fibrosis. Nat. Med. 2006, 12, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 1998, 82, 1131–1149. [Google Scholar] [CrossRef]
- Auguet, T.; Berlanga, A.; Guiu-Jurado, E.; Terra, X.; Martinez, S.; Aguilar, C.; Filiu, E.; Alibalic, A.; Sabench, F.; Hernández, M.; et al. Endocannabinoid receptors gene expression in morbidly obese women with nonalcoholic fatty liver disease. BioMed Res. Int. 2014, 2014, 502542. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef]
- Mallat, A.; Teixeira-Clerc, F.; Deveaux, V.; Manin, S.; Lotersztajn, S. The endocannabinoid system as a key mediator during liver diseases: New insights and therapeutic openings. Br. J. Pharmacol. 2011, 163, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Mallat, A.; Teixeira-Clerc, F.; Lotersztajn, S. Cannabinoid signaling and liver therapeutics. J. Hepatol. 2013, 59, 891–896. [Google Scholar] [CrossRef]
- Alswat, K.A. The role of endocannabinoids system in fatty liver disease and therapeutic potentials. Saudi J. Gastroenterol. 2013, 19, 144–151. [Google Scholar] [CrossRef]
- Deveaux, V.; Cadoudal, T.; Ichigotani, Y.; Teixeira-Clerc, F.; Louvet, A.; Manin, S.; Nhieu, J.T.-V.; Belot, M.P.; Zimmer, A.; Even, P.; et al. Cannabinoid CB2 receptor potentiates obesity-associated inflammation, insulin resistance and hepatic steatosis. PLoS ONE 2009, 4, e5844. [Google Scholar] [CrossRef]
- González-Mariscal, I.; Krzysik-Walker, S.M.; Doyle, M.E.; Liu, Q.-R.; Cimbro, R.; Santa-Cruz Calvo, S.; Ghosh, S.; Cieśla, Ł.; Moaddel, R.; Carlson, O.D.; et al. Human CB1 Receptor Isoforms, present in Hepatocytes and β-cells, are Involved in Regulating Metabolism. Sci. Rep. 2016, 6, 33302. [Google Scholar]
- Godlewski, G.; Kunos, G. Overview of Nonclassical Cannabinoid Receptors. In The Receptors; Abood, M., Sorensen, R., Stella, N., Eds.; Springer: New York, NY, USA, 2013; Volume 24. [Google Scholar]
- Nie, J.; Lewis, D.L. The proximal and distal C-terminal tail domains of the CB1 cannabinoid receptor mediate G protein coupling. Neuroscience 2001, 107, 161–167. [Google Scholar] [CrossRef]
- Nie, J.; Lewis, D.L. Structural domains of the CB1 cannabinoid receptor that contribute to constitutive activity and G-protein sequestration. J. Neurosci. 2001, 21, 8758–8764. [Google Scholar] [CrossRef]
- Stadel, R.; Ahn, K.H.; Kendall, D.A. The cannabinoid type-1 receptor carboxyl-terminus, more than just a tail. J. Neurochem. 2011, 117, 1–18. [Google Scholar] [CrossRef]
- Bazwinsky-Wutschke, I.; Zipprich, A.; Dehghani, F. Daytime-Dependent Changes of Cannabinoid Receptor Type 1 and Type 2 Expression in Rat Liver. Int. J. Mol. Sci. 2017, 18, 1844. [Google Scholar] [CrossRef]
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458. [Google Scholar] [CrossRef]
- Chanda, D.; Kim, D.-K.; Li, T.; Kim, Y.-H.; Koo, S.-H.; Lee, C.-H.; Chiang, J.Y.L.; Choi, H.-S. Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J. Biol. Chem. 2011, 286, 27971–27979. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, H.; Qiu, Y.; Chen, X.; Mendez, R.; Dandekar, A.; Zhang, X.; Zhang, C.; Liu, A.C.; Yin, L.; et al. CREBH Couples Circadian Clock With Hepatic Lipid Metabolism. Diabetes 2016, 65, 3369–3383. [Google Scholar] [CrossRef]
- Chanda, D.; Kim, Y.-H.; Kim, D.-K.; Lee, M.-W.; Lee, S.-Y.; Park, T.-S.; Koo, S.-H.; Lee, C.-H.; Choi, H.-S. Activation of cannabinoid receptor type 1 (Cb1r) disrupts hepatic insulin receptor signaling via cyclic AMP-response element-binding protein H (Crebh)-mediated induction of Lipin1 gene. J. Biol. Chem. 2012, 287, 38041–38049. [Google Scholar] [CrossRef]
- Lamia, K.A.; Storch, K.-F.; Weitz, C.J. Physiological significance of a peripheral tissue circadian clock. Proc. Natl. Acad. Sci. USA 2008, 105, 15172–15177. [Google Scholar] [CrossRef]
- Fukuda, H.; Iritani, N. Diurnal variations of lipogenic enzyme mRNA quantities in rat liver. Biochim. Biophys. Acta 1991, 1086, 261–264. [Google Scholar] [CrossRef]
- Zardoya, R.; Diez, A.; Serradilla, M.C.; Madrid, J.A.; Bautista, J.M.; Garrido-Pertierra, A. Lipogenic activities in rat liver are subjected to circadian rhythms. Rev. Esp. De Fisiol. 1994, 50, 239–244. [Google Scholar]
- Kunos, G. Understanding metabolic homeostasis and imbalance: What is the role of the endocannabinoid system? Am. J. Med. 2007, 120, S18–S24. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cinar, R.; Xiong, K.; Godlewski, G.; Jourdan, T.; Lin, Y.; Ntambi, J.M.; Kunos, G. Monounsaturated fatty acids generated via stearoyl CoA desaturase-1 are endogenous inhibitors of fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2013, 110, 18832–18837. [Google Scholar] [CrossRef] [PubMed]
- Murdolo, G.; Kempf, K.; Hammarstedt, A.; Herder, C.; Smith, U.; Jansson, P.-A. Insulin differentially modulates the peripheral endocannabinoid system in human subcutaneous abdominal adipose tissue from lean and obese individuals. J. Endocrinol. Investig. 2007, 30, RC17–RC21. [Google Scholar] [CrossRef] [PubMed]
- Ravinet Trillou, C.; Arnone, M.; Delgorge, C.; Gonalons, N.; Keane, P.; Maffrand, J.-P.; Soubrie, P. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R345–R353. [Google Scholar] [CrossRef]
- Cinar, R.; Godlewski, G.; Liu, J.; Tam, J.; Jourdan, T.; Mukhopadhyay, B.; Harvey-White, J.; Kunos, G. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology 2014, 59, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Demizieux, L.; Gresti, J.; Djaouti, L.; Gaba, L.; Vergès, B.; Degrace, P. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: Evidence from cultured explants. Hepatology 2012, 55, 790–799. [Google Scholar] [CrossRef]
- Nogueiras, R.; Veyrat-Durebex, C.; Suchanek, P.M.; Klein, M.; Tschöp, J.; Caldwell, C.; Woods, S.C.; Wittmann, G.; Watanabe, M.; Liposits, Z.; et al. Peripheral, but not central, CB1 antagonism provides food intake-independent metabolic benefits in diet-induced obese rats. Diabetes 2008, 57, 2977–2991. [Google Scholar] [CrossRef]
- de Gottardi, A.; Spahr, L.; Ravier-Dall’Antonia, F.; Hadengue, A. Cannabinoid receptor 1 and 2 agonists increase lipid accumulation in hepatocytes. Liver Int. 2010, 30, 1482–1489. [Google Scholar] [CrossRef]
- Chen, J.; Montagner, A.; Tan, N.S.; Wahli, W. Insights into the Role of PPARβ/δ in NAFLD. Int. J. Mol. Sci. 2018, 19, 1893. [Google Scholar] [CrossRef]
- Wu, H.M.; Yang, Y.M.; Kim, S.G. Rimonabant, a cannabinoid receptor type 1 inverse agonist, inhibits hepatocyte lipogenesis by activating liver kinase B1 and AMP-activated protein kinase axis downstream of Gα i/o inhibition. Mol. Pharmacol. 2011, 80, 859–869. [Google Scholar] [CrossRef]
- Tam, J.; Vemuri, V.K.; Liu, J.; Bátkai, S.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.V.; Pickel, J.; et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Investig. 2010, 120, 2953–2966. [Google Scholar] [CrossRef]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.-S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Gary-Bobo, M.; Elachouri, G.; Gallas, J.F.; Janiak, P.; Marini, P.; Ravinet-Trillou, C.; Chabbert, M.; Cruccioli, N.; Pfersdorff, C.; Roque, C.; et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology 2007, 46, 122–129. [Google Scholar] [CrossRef]
- Jourdan, T.; Djaouti, L.; Demizieux, L.; Gresti, J.; Vergès, B.; Degrace, P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes 2010, 59, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 2012, 142, 1218–1228. [Google Scholar] [CrossRef]
- Ruby, M.A.; Nomura, D.K.; Hudak, C.S.S.; Mangravite, L.M.; Chiu, S.; Casida, J.E.; Krauss, R.M. Overactive endocannabinoid signaling impairs apolipoprotein E-mediated clearance of triglyceride-rich lipoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 14561–14566. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.-I.; Bátkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef]
- Lipina, C.; Vaanholt, L.M.; Davidova, A.; Mitchell, S.E.; Storey-Gordon, E.; Hambly, C.; Irving, A.J.; Speakman, J.R.; Hundal, H.S. CB1 receptor blockade counters age-induced insulin resistance and metabolic dysfunction. Aging Cell 2016, 15, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Agudo, J.; Martin, M.; Roca, C.; Molas, M.; Bura, A.S.; Zimmer, A.; Bosch, F.; Maldonado, R. Deficiency of CB2 cannabinoid receptor in mice improves insulin sensitivity but increases food intake and obesity with age. Diabetologia 2010, 53, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, F.H.; Lefebvre, P.J.; Scheen, A.J. Non-alcoholic steatohepatitis: Association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab. 2000, 26, 98–106. [Google Scholar]
- Yki-Järvinen, H. Fat in the liver and insulin resistance. Ann. Med. 2005, 37, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Chorvat, R.J. Peripherally restricted CB1 receptor blockers. Bioorganic Med. Chem. Lett. 2013, 23, 4751–4760. [Google Scholar] [CrossRef]
- Banasch, M.; Goetze, O.; Schmidt, W.E.; Meier, J.J. Rimonabant as a novel therapeutic option for nonalcoholic steatohepatitis. Liver Int. 2007, 27, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J.; Paquot, N. Inhibitors of cannabinoid receptors and glucose metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 505–511. [Google Scholar] [CrossRef]
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The cannabinoid receptor CB1 modulates the signaling properties of the lysophosphatidylinositol receptor GPR55. J. Biol. Chem. 2012, 287, 44234–44248. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharmacol. 2007, 152, 984–986. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Moriconi, A.; Cerbara, I.; Maccarrone, M.; Topai, A. GPR55: Current knowledge and future perspectives of a purported “Type-3” cannabinoid receptor. Curr. Med. Chem. 2010, 17, 1411–1429. [Google Scholar] [CrossRef]
- Petitet, F.; Donlan, M.; Michel, A. GPR55 as a new cannabinoid receptor: Still a long way to prove it. Chem. Biol. Drug Des. 2006, 67, 252–253. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Abood, M.E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther. 2010, 126, 301–313. [Google Scholar] [CrossRef]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Catalán, V.; Whyte, L.; Díaz-Arteaga, A.; Vázquez-Martínez, R.; Rotellar, F.; Guzmá, R.; Gómez-Ambrosi, J.; Pulido, M.R.; Russell, W.R.; et al. The L-α-lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes 2012, 61, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Simcocks, A.C.; O’Keefe, L.; Jenkin, K.A.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. A potential role for GPR55 in the regulation of energy homeostasis. Drug Discov. Today 2014, 19, 1145–1151. [Google Scholar] [CrossRef]
- Oka, S.; Toshida, T.; Maruyama, K.; Nakajima, K.; Yamashita, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem. 2009, 145, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Masquelier, J.; Muccioli, G.G. Development and validation of a specific and sensitive HPLC-ESI-MS method for quantification of lysophosphatidylinositols and evaluation of their levels in mice tissues. J. Pharm. Biomed. Anal. 2016, 126, 132–140. [Google Scholar] [CrossRef]
- Lipina, C.; Walsh, S.K.; Mitchell, S.E.; Speakman, J.R.; Wainwright, C.L.; Hundal, H.S. GPR55 deficiency is associated with increased adiposity and impaired insulin signaling in peripheral metabolic tissues. FASEB J. 2018, 33, 1299–1312. [Google Scholar] [CrossRef]
- Díaz-Arteaga, A.; Vázquez, M.J.; Vazquez-Martínez, R.; Pulido, M.R.; Suarez, J.; Velásquez, D.A.; López, M.; Ross, R.A.; de Fonseca, F.R.; Bermudez-Silva, F.J.; et al. The atypical cannabinoid O-1602 stimulates food intake and adiposity in rats. Diabetes Obes. Metab. 2012, 14, 234–243. [Google Scholar]
- Balenga, N.A.B.; Aflaki, E.; Kargl, J.; Platzer, W.; Schröder, R.; Blättermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef]
- Ning, Y.; O’Neill, K.; Lan, H.; Pang, L.; Shan, L.X.; Hawes, B.E.; Hedrick, J.A. Endogenous and synthetic agonists of GPR119 differ in signalling pathways and their effects on insulin secretion in MIN6c4 insulinoma cells. Br. J. Pharmacol. 2008, 155, 1056–1065. [Google Scholar] [CrossRef]
- Moran, B.M.; Flatt, P.R.; McKillop, A.M. G protein-coupled receptors: Signalling and regulation by lipid agonists for improved glucose homoeostasis. Acta Diabetol. 2016, 53, 177–188. [Google Scholar] [CrossRef]
- Yang, J.W.; Kim, H.S.; Im, J.H.; Kim, J.W.; Jun, D.W.; Lim, S.C.; Lee, K.; Choi, J.M.; Kim, S.K.; Kang, K.W. GPR119: A promising target for nonalcoholic fatty liver disease. FASEB J. 2016, 30, 324–335. [Google Scholar] [CrossRef]
- Yang, J.W.; Kim, H.S.; Choi, Y.-W.; Kim, Y.-M.; Kang, K.W. Therapeutic application of GPR119 ligands in metabolic disorders. Diabetes Obes. Metab. 2018, 20, 257–269. [Google Scholar] [CrossRef]
- Bahirat, U.A.; Shenoy, R.R.; Goel, R.N.; Nemmani, K.V.S. APD668, a G protein-coupled receptor 119 agonist improves fat tolerance and attenuates fatty liver in high-trans fat diet induced steatohepatitis model in C57BL/6 mice. Eur. J. Pharmacol. 2017, 801, 35–45. [Google Scholar] [CrossRef]
- Bahirat, U.A.; Shenoy, R.R.; Talwar, R.; Goel, R.N.; Nemmani, K.V.S. Co-administration of APD668, a G protein-coupled receptor 119 agonist and linagliptin, a DPPIV inhibitor, prevents progression of steatohepatitis in mice fed on a high trans-fat diet. Biochem. Biophys. Res. Commun. 2018, 495, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Pistis, M.; Melis, M. From surface to nuclear receptors: The endocannabinoid family extends its assets. Curr. Med. Chem. 2010, 17, 1450–1467. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Zardi, E.M.; Navarini, L.; Sambataro, G.; Piccinni, P.; Sambataro, F.M.; Spina, C.; Dobrina, A. Hepatic PPARs: Their role in liver physiology, fibrosis and treatment. Curr. Med. Chem. 2013, 20, 3370–3396. [Google Scholar] [CrossRef] [PubMed]
- Charbonnel, B. PPAR-alpha and PPAR-gamma agonists for type 2 diabetes. Lancet 2009, 374, 96–98. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.-C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef]
- Tailleux, A.; Wouters, K.; Staels, B. Roles of PPARs in NAFLD: Potential therapeutic targets. Biochim. Biophys. Acta 2012, 1821, 809–818. [Google Scholar] [CrossRef]
- Peters, J.M.; Rusyn, I.; Rose, M.L.; Gonzalez, F.J.; Thurman, R.G. Peroxisome proliferator-activated receptor alpha is restricted to hepatic parenchymal cells, not Kupffer cells: Implications for the mechanism of action of peroxisome proliferators in hepatocarcinogenesis. Carcinogenesis 2000, 21, 823–826. [Google Scholar] [CrossRef]
- Hoekstra, M.; Kruijt, J.K.; van Eck, M.; van Berkel, T.J.C. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J. Biol. Chem. 2003, 278, 25448–25453. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Régnier, M.; Polizzi, A.; Lippi, Y.; Fouché, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the role of hepatocyte PPARα activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Gervois, P.; Kleemann, R.; Pilon, A.; Percevault, F.; Koenig, W.; Staels, B.; Kooistra, T. Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J. Biol. Chem. 2004, 279, 16154–16160. [Google Scholar] [CrossRef]
- Toyama, T.; Nakamura, H.; Harano, Y.; Yamauchi, N.; Morita, A.; Kirishima, T.; Minami, M.; Itoh, Y.; Okanoue, T. PPARalpha ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochem. Biophys. Res. Commun. 2004, 324, 697–704. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Fu, J.; Oveisi, F.; Gaetani, S.; Lin, E.; Piomelli, D. Oleoylethanolamide, an endogenous PPAR-alpha agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 2005, 48, 1147–1153. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J.; Qiu, Y. Oleoylethanolamide, an endogenous PPAR-α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef]
- Costet, P.; Legendre, C.; Moré, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: No alteration in adipose tissue of obese and NIDDM patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef]
- Tugwood, J.D.; Aldridge, T.C.; Lambe, K.G.; Macdonald, N.; Woodyatt, N.J. Peroxisome proliferator-activated receptors: Structures and function. Ann. N. Y. Acad. Sci. 1996, 804, 252–265. [Google Scholar] [CrossRef]
- Mukherjee, R.; Jow, L.; Croston, G.E.; Paterniti, J.R. Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J. Biol. Chem. 1997, 272, 8071–8076. [Google Scholar] [CrossRef]
- Girroir, E.E.; Hollingshead, H.E.; He, P.; Zhu, B.; Perdew, G.H.; Peters, J.M. Quantitative expression patterns of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) protein in mice. Biochem. Biophys. Res. Commun. 2008, 371, 456–461. [Google Scholar] [CrossRef]
- Kostadinova, R.; Montagner, A.; Gouranton, E.; Fleury, S.; Guillou, H.; Dombrowicz, D.; Desreumaux, P.; Wahli, W. GW501516-activated PPARβ/δ promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012, 2, 34. [Google Scholar] [CrossRef]
- Lee, C.-H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.-W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Müller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol. Genom. 2010, 41, 42–52. [Google Scholar] [CrossRef]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.-C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [PubMed]
- Boelsterli, U.A.; Bedoucha, M. Toxicological consequences of altered peroxisome proliferator-activated receptor gamma (PPARgamma) expression in the liver: Insights from models of obesity and type 2 diabetes. Biochem. Pharmacol. 2002, 63, 1–10. [Google Scholar] [CrossRef]
- Rahimian, R.; Masih-Khan, E.; Lo, M.; van Breemen, C.; McManus, B.M.; Dubé, G.P. Hepatic over-expression of peroxisome proliferator activated receptor gamma2 in the ob/ob mouse model of non-insulin dependent diabetes mellitus. Mol. Cell. Biochem. 2001, 224, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Memon, R.A.; Tecott, L.H.; Nonogaki, K.; Beigneux, A.; Moser, A.H.; Grunfeld, C.; Feingold, K.R. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: Troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000, 141, 4021–4031. [Google Scholar] [PubMed]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clària, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Schadinger, S.E.; Bucher, N.L.R.; Schreiber, B.M.; Farmer, S.R. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1195–E1205. [Google Scholar] [CrossRef]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Sirén, J.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef]
- Pettinelli, P.; Videla, L.A. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: An additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.-H.; Gavrilova, O.; Ward, J.M.; Brewer, B.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef]
- Matsusue, K.; Aibara, D.; Hayafuchi, R.; Matsuo, K.; Takiguchi, S.; Gonzalez, F.J.; Yamano, S. Hepatic PPARγ and LXRα independently regulate lipid accumulation in the livers of genetically obese mice. FEBS Lett. 2014, 588, 2277–2281. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Hernandez-Ono, A.; Siri, P.; Weisberg, S.; Conlon, D.; Graham, M.J.; Crooke, R.M.; Huang, L.-S.; Ginsberg, H.N. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J. Biol. Chem. 2006, 281, 37603–37615. [Google Scholar] [CrossRef] [PubMed]
- Wolf Greenstein, A.; Majumdar, N.; Yang, P.; Subbaiah, P.V.; Kineman, R.D.; Cordoba-Chacon, J. Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2017, 232, 107–121. [Google Scholar] [CrossRef]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.-S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef]
- Noureddin, M.; Anstee, Q.M.; Loomba, R. Review article: Emerging anti-fibrotic therapies in the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2016, 43, 1109–1123. [Google Scholar] [CrossRef]
- Saile, B.; Ramadori, G. Inflammation, damage repair and liver fibrosis--role of cytokines and different cell types. Z. Gastroenterol. 2007, 45, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, S.V.; Schwabe, R.F. Endocannabinoids and liver disease. II. Endocannabinoids in the pathogenesis and treatment of liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G357–G362. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef]
- Kunos, G.; Osei-Hyiaman, D.; Bátkai, S.; Sharkey, K.A.; Makriyannis, A. Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol. Sci. 2009, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.-I.; Osei-Hyiaman, D.; Park, O.; Liu, J.; Bátkai, S.; Mukhopadhyay, P.; Horiguchi, N.; Harvey-White, J.; Marsicano, G.; Lutz, B.; et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008, 7, 227–235. [Google Scholar] [CrossRef]
- Izzo, A.A.; Piscitelli, F.; Capasso, R.; Aviello, G.; Romano, B.; Borrelli, F.; Petrosino, S.; Di Marzo, V. Peripheral endocannabinoid dysregulation in obesity: Relation to intestinal motility and energy processing induced by food deprivation and re-feeding. Br. J. Pharmacol. 2009, 158, 451–461. [Google Scholar] [CrossRef]
- Gabbay, E.; Avraham, Y.; Ilan, Y.; Israeli, E.; Berry, E.M. Endocannabinoids and liver disease--review. Liver Int. 2005, 25, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Azar, S.; Nemirovski, A.; Webb, M.; Halpern, Z.; Shibolet, O.; Tam, J. Serum levels of endocannabinoids are independently associated with nonalcoholic fatty liver disease. Obesity 2017, 25, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, P.; Viola, A.; Piscitelli, F.; Giannone, F.; Berzigotti, A.; Cescon, M.; Domenicali, M.; Petrosino, S.; Giampalma, E.; Riili, A.; et al. Circulating and hepatic endocannabinoids and endocannabinoid-related molecules in patients with cirrhosis. Liver Int. 2010, 30, 816–825. [Google Scholar] [CrossRef]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Uchinami, H.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. Anandamide induces necrosis in primary hepatic stellate cells. Hepatology 2005, 41, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Liu, J.; Mukhopadhyay, B.; Cinar, R.; Godlewski, G.; Kunos, G. Endocannabinoids in liver disease. Hepatology 2011, 53, 346–355. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Qian, T.; de Minicis, S.; Harvey-White, J.; Kunos, G.; Vinod, K.Y.; Hungund, B.; Schwabe, R.F. The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB J. 2007, 21, 2798–2806. [Google Scholar] [CrossRef]
- Mackie, K.; Stella, N. Cannabinoid receptors and endocannabinoids: Evidence for new players. AAPS J. 2006, 8, E298–E306. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V. Cannabinoid receptors and the regulation of immune response. Chem. Phys. Lipids 2000, 108, 169–190. [Google Scholar] [CrossRef]
- Ross, R.A.; Brockie, H.C.; Pertwee, R.G. Inhibition of nitric oxide production in RAW264.7 macrophages by cannabinoids and palmitoylethanolamide. Eur. J. Pharmacol. 2000, 401, 121–130. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Kishimoto, S.; Miyashita, T.; Nakane, S.; Kodaka, T.; Suhara, Y.; Takayama, H.; Waku, K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 2000, 275, 605–612. [Google Scholar] [CrossRef]
- Lambert, D.M.; Vandevoorde, S.; Jonsson, K.-O.; Fowler, C.J. The palmitoylethanolamide family: A new class of anti-inflammatory agents? Curr. Med. Chem. 2002, 9, 663–674. [Google Scholar] [CrossRef]
- Wojtalla, A.; Herweck, F.; Granzow, M.; Klein, S.; Trebicka, J.; Huss, S.; Lerner, R.; Lutz, B.; Schildberg, F.A.; Knolle, P.A.; et al. The endocannabinoid N-arachidonoyl dopamine (NADA) selectively induces oxidative stress-mediated cell death in hepatic stellate cells but not in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G873–G887. [Google Scholar] [CrossRef]
- Hézode, C.; Roudot-Thoraval, F.; Nguyen, S.; Grenard, P.; Julien, B.; Zafrani, E.-S.; Pawlotsky, J.-M.; Pawlostky, J.-M.; Dhumeaux, D.; Lotersztajn, S.; et al. Daily cannabis smoking as a risk factor for progression of fibrosis in chronic hepatitis C. Hepatology 2005, 42, 63–71. [Google Scholar]
- Bátkai, S.; Járai, Z.; Wagner, J.A.; Goparaju, S.K.; Varga, K.; Liu, J.; Wang, L.; Mirshahi, F.; Khanolkar, A.D.; Makriyannis, A.; et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat. Med. 2001, 7, 827–832. [Google Scholar]
- Baldassarre, M.; Giannone, F.A.; Napoli, L.; Tovoli, A.; Ricci, C.S.; Tufoni, M.; Caraceni, P. The endocannabinoid system in advanced liver cirrhosis: Pathophysiological implication and future perspectives. Liver Int. 2013, 33, 1298–1308. [Google Scholar] [CrossRef]
- Trebicka, J.; Racz, I.; Siegmund, S.V.; Cara, E.; Granzow, M.; Schierwagen, R.; Klein, S.; Wojtalla, A.; Hennenberg, M.; Huss, S.; et al. Role of cannabinoid receptors in alcoholic hepatic injury: Steatosis and fibrogenesis are increased in CB2 receptor-deficient mice and decreased in CB1 receptor knockouts. Liver Int. 2011, 31, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Mallat, A.; Lotersztajn, S. Endocannabinoids as novel mediators of liver diseases. J. Endocrinol. Investig. 2006, 29, 58–65. [Google Scholar]
- Yang, Y.-Y.; Lin, H.-C.; Huang, Y.-T.; Lee, T.-Y.; Hou, M.-C.; Wang, Y.-W.; Lee, F.-Y.; Lee, S.-D. Effect of chronic CB1 cannabinoid receptor antagonism on livers of rats with biliary cirrhosis. Clin. Sci. 2007, 112, 533–542. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Kanel, G.C.; Atkinson, R.D.; McCuskey, R.S. Prevention of hepatic fibrosis in a murine model of metabolic syndrome with nonalcoholic steatohepatitis. Am. J. Pathol. 2008, 173, 993–1001. [Google Scholar] [CrossRef]
- Domenicali, M.; Caraceni, P.; Giannone, F.; Pertosa, A.M.; Principe, A.; Zambruni, A.; Trevisani, F.; Croci, T.; Bernardi, M. Cannabinoid type 1 receptor antagonism delays ascites formation in rats with cirrhosis. Gastroenterology 2009, 137, 341–349. [Google Scholar] [CrossRef]
- Giannone, F.A.; Baldassarre, M.; Domenicali, M.; Zaccherini, G.; Trevisani, F.; Bernardi, M.; Caraceni, P. Reversal of liver fibrosis by the antagonism of endocannabinoid CB1 receptor in a rat model of CCl(4)-induced advanced cirrhosis. Lab. Investig. 2012, 92, 384–395. [Google Scholar] [CrossRef]
- Mukhopadhyay, B.; Cinar, R.; Yin, S.; Liu, J.; Tam, J.; Godlewski, G.; Harvey-White, J.; Mordi, I.; Cravatt, B.F.; Lotersztajn, S.; et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc. Natl. Acad. Sci. USA 2011, 108, 6323–6328. [Google Scholar] [CrossRef]
- Teixeira-Clerc, F.; Belot, M.-P.; Manin, S.; Deveaux, V.; Cadoudal, T.; Chobert, M.-N.; Louvet, A.; Zimmer, A.; Tordjmann, T.; Mallat, A.; et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology 2010, 52, 1046–1059. [Google Scholar] [CrossRef]
- Muñoz-Luque, J.; Ros, J.; Fernández-Varo, G.; Tugues, S.; Morales-Ruiz, M.; Alvarez, C.E.; Friedman, S.L.; Arroyo, V.; Jiménez, W. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J. Pharmacol. Exp. Ther. 2008, 324, 475–483. [Google Scholar] [CrossRef]
- Reichenbach, V.; Ros, J.; Fernández-Varo, G.; Casals, G.; Melgar-Lesmes, P.; Campos, T.; Makriyannis, A.; Morales-Ruiz, M.; Jiménez, W. Prevention of fibrosis progression in CCl4-treated rats: Role of the hepatic endocannabinoid and apelin systems. J. Pharmacol. Exp. Ther. 2012, 340, 629–637. [Google Scholar] [CrossRef]
- Guillot, A.; Hamdaoui, N.; Bizy, A.; Zoltani, K.; Souktani, R.; Zafrani, E.-S.; Mallat, A.; Lotersztajn, S.; Lafdil, F. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology 2014, 59, 296–306. [Google Scholar] [CrossRef]
- Rossi, F.; Bellini, G.; Alisi, A.; Alterio, A.; Maione, S.; Perrone, L.; Locatelli, F.; Miraglia del Giudice, E.; Nobili, V. Cannabinoid receptor type 2 functional variant influences liver damage in children with non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e42259. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef]
- Knittel, T.; Kobold, D.; Saile, B.; Grundmann, A.; Neubauer, K.; Piscaglia, F.; Ramadori, G. Rat liver myofibroblasts and hepatic stellate cells: Different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology 1999, 117, 1205–1221. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Shah, V.; Rockey, D.C. Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J. Hepatol. 2014, 61, 912–924. [Google Scholar] [CrossRef]
- Bernardi, M.; Moreau, R.; Angeli, P.; Schnabl, B.; Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 2015, 63, 1272–1284. [Google Scholar] [CrossRef]
- Wiest, R.; Groszmann, R.J. The paradox of nitric oxide in cirrhosis and portal hypertension: Too much, not enough. Hepatology 2002, 35, 478–491. [Google Scholar] [CrossRef]
- Bosch, J.; Groszmann, R.J.; Shah, V.H. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J. Hepatol. 2015, 62, S121–S130. [Google Scholar] [CrossRef]
- Moezi, L.; Gaskari, S.A.; Liu, H.; Baik, S.K.; Dehpour, A.R.; Lee, S.S. Anandamide mediates hyperdynamic circulation in cirrhotic rats via CB(1) and VR(1) receptors. Br. J. Pharmacol. 2006, 149, 898–908. [Google Scholar] [CrossRef]
- Varga, K.; Wagner, J.A.; Bridgen, D.T.; Kunos, G. Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB J. 1998, 12, 1035–1044. [Google Scholar] [CrossRef]
- Liu, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Wagner, J.A.; Cravatt, B.F.; Gao, B.; Kunos, G. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J. Biol. Chem. 2003, 278, 45034–45039. [Google Scholar] [CrossRef]
- Domenicali, M.; Ros, J.; Fernández-Varo, G.; Cejudo-Martín, P.; Crespo, M.; Morales-Ruiz, M.; Briones, A.M.; Campistol, J.-M.; Arroyo, V.; Vila, E.; et al. Increased anandamide induced relaxation in mesenteric arteries of cirrhotic rats: Role of cannabinoid and vanilloid receptors. Gut 2005, 54, 522–527. [Google Scholar] [CrossRef]
- Yang, Y.-Y.; Lin, H.-C. Alteration of intrahepatic microcirculation in cirrhotic livers. J. Chin. Med Assoc. 2015, 78, 430–437. [Google Scholar] [CrossRef][Green Version]
- Orliac, M.L.; Peroni, R.; Celuch, S.M.; Adler-Graschinsky, E. Potentiation of anandamide effects in mesenteric beds isolated from endotoxemic rats. J. Pharmacol. Exp. Ther. 2003, 304, 179–184. [Google Scholar] [CrossRef]
- Yotti, R.; Ripoll, C.; Benito, Y.; Catalina, M.V.; Elízaga, J.; Rincón, D.; Fernández-Avilés, F.; Bermejo, J.; Bañares, R. Left ventricular systolic function is associated with sympathetic nervous activity and markers of inflammation in cirrhosis. Hepatology 2017, 65, 2019–2030. [Google Scholar] [CrossRef]
- Gebremedhin, D.; Lange, A.R.; Campbell, W.B.; Hillard, C.J.; Harder, D.R. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am. J. Physiol. 1999, 276, H2085–H2093. [Google Scholar] [CrossRef]
- Bátkai, S.; Mukhopadhyay, P.; Harvey-White, J.; Kechrid, R.; Pacher, P.; Kunos, G. Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in vivo in cirrhotic rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1689–H1695. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazwinsky-Wutschke, I.; Zipprich, A.; Dehghani, F. Endocannabinoid System in Hepatic Glucose Metabolism, Fatty Liver Disease, and Cirrhosis. Int. J. Mol. Sci. 2019, 20, 2516. https://doi.org/10.3390/ijms20102516
Bazwinsky-Wutschke I, Zipprich A, Dehghani F. Endocannabinoid System in Hepatic Glucose Metabolism, Fatty Liver Disease, and Cirrhosis. International Journal of Molecular Sciences. 2019; 20(10):2516. https://doi.org/10.3390/ijms20102516
Chicago/Turabian StyleBazwinsky-Wutschke, Ivonne, Alexander Zipprich, and Faramarz Dehghani. 2019. "Endocannabinoid System in Hepatic Glucose Metabolism, Fatty Liver Disease, and Cirrhosis" International Journal of Molecular Sciences 20, no. 10: 2516. https://doi.org/10.3390/ijms20102516
APA StyleBazwinsky-Wutschke, I., Zipprich, A., & Dehghani, F. (2019). Endocannabinoid System in Hepatic Glucose Metabolism, Fatty Liver Disease, and Cirrhosis. International Journal of Molecular Sciences, 20(10), 2516. https://doi.org/10.3390/ijms20102516