Senescence Phenomena and Metabolic Alteration in Mesenchymal Stromal Cells from a Mouse Model of Rett Syndrome


 ,
, 
 ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
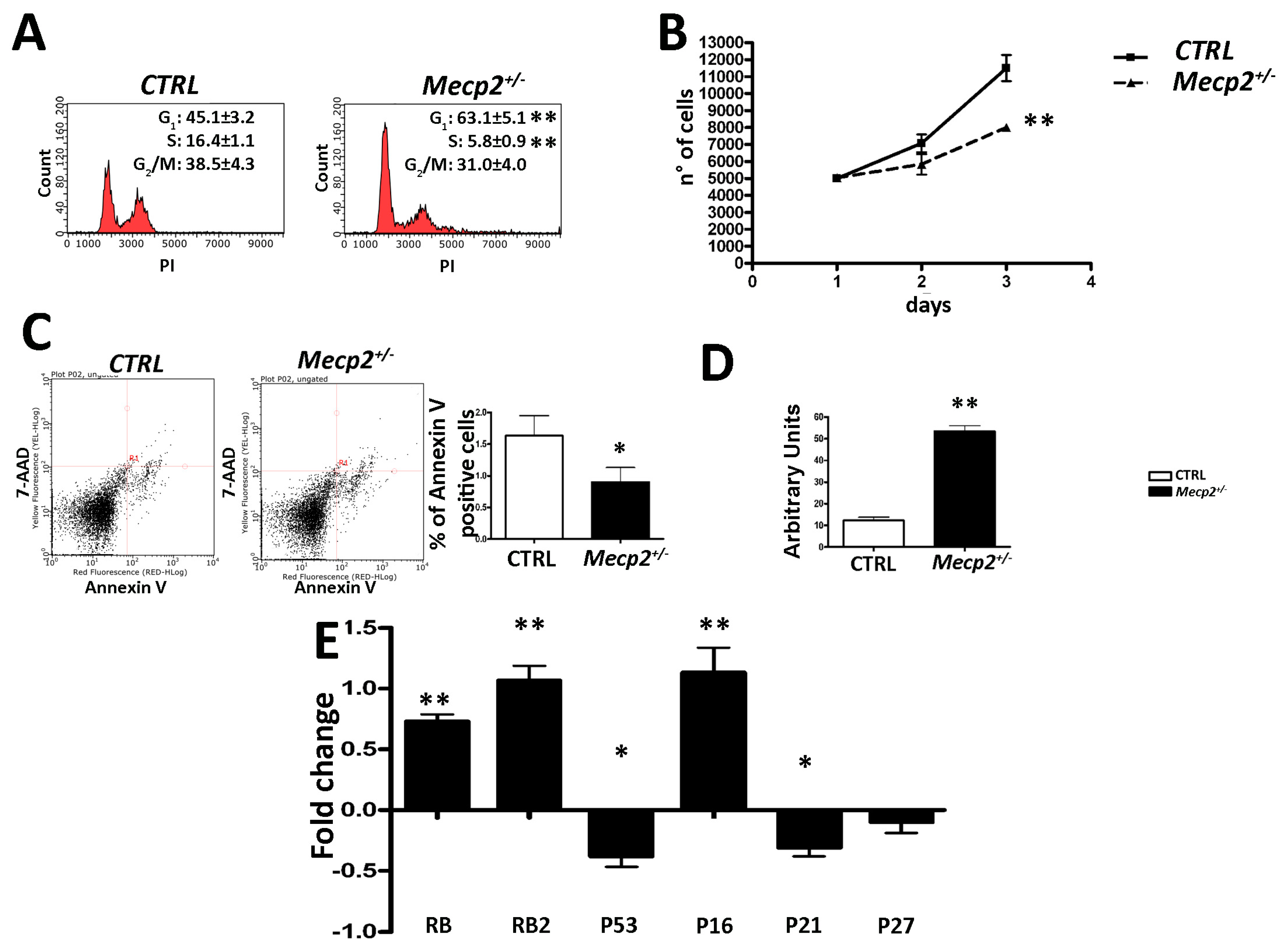
2.1. MSCs from Mecp2+/− Mice Showed a Lower Degree of Proliferation and Apoptosis and Are Prone to Senescence
2.2. Impaired DNA Repair System in MSCs from Mecp2+/− Mice
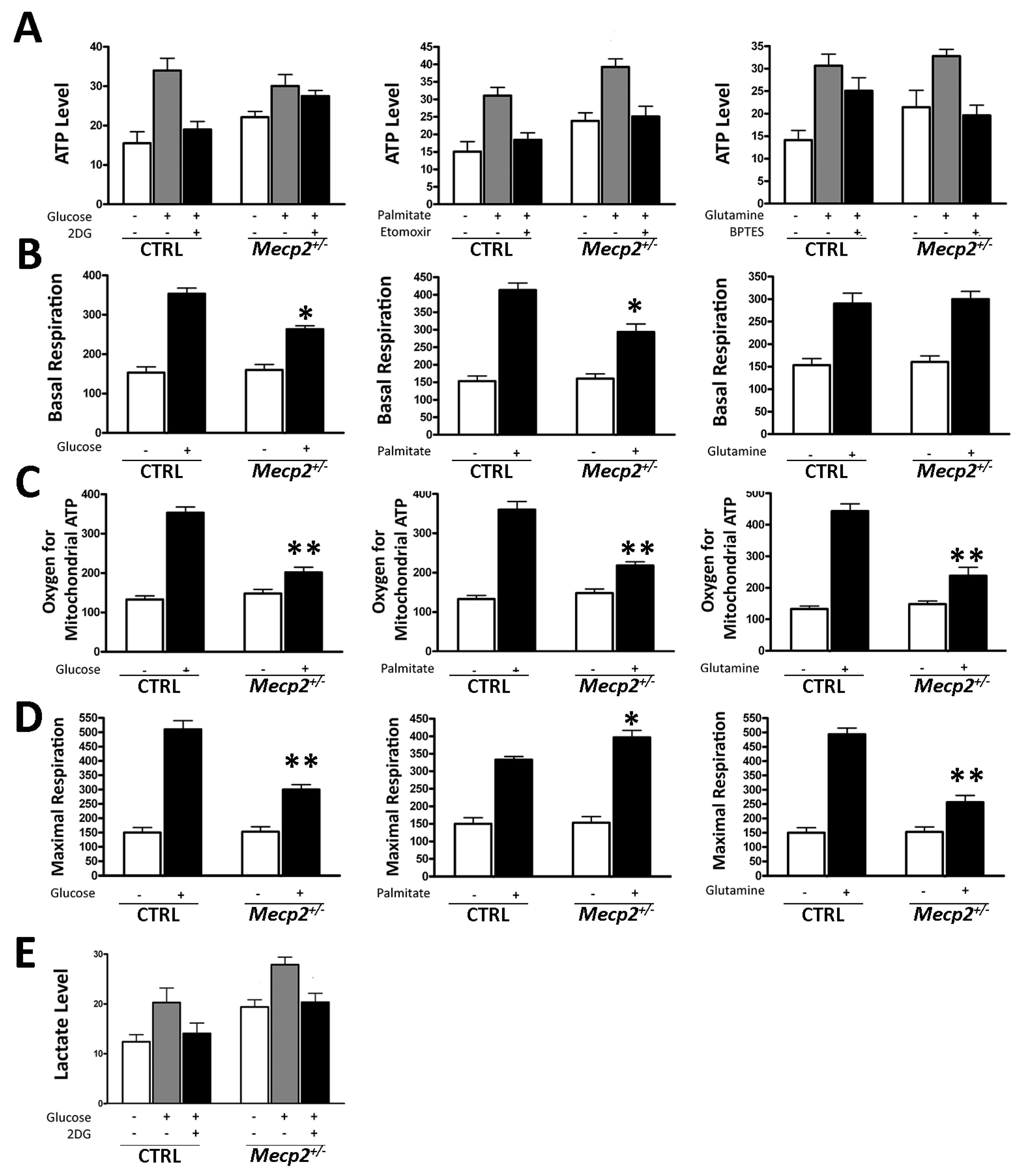
2.3. Mecp2+/− MSCs Exhibited Metabolic Flexibility but Mitochondrial Energy Production Impairment
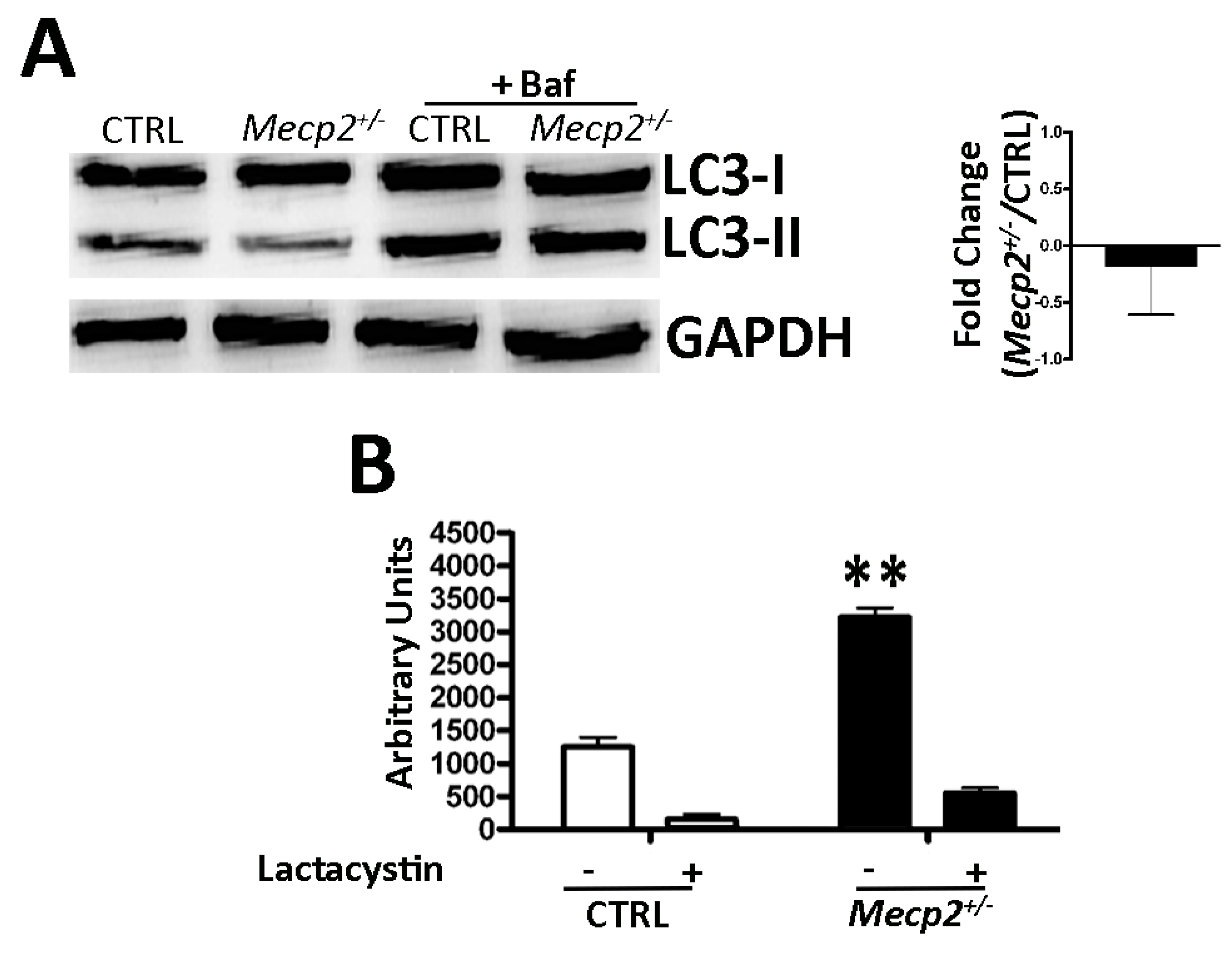
2.4. Autophagy Was Not Impaired in Mecp2+/− MSCs
2.5. Mecp2+/− Mice Showed an Increase in Proteasome Activity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Genotyping
4.3. Mouse MSC Cultures
4.4. Cell Cycle Analysis
4.5. Proliferation Analysis
4.6. Senescence Detection
4.7. Apoptosis Detection
4.8. Necrosis Detection
4.9. RNA Extraction and RT-qPCR
4.10. Treatment with Hydrogen Peroxide, Doxorubicin, and Ultraviolet Irradiation
4.11. Immunocytochemistry for γ-H2AX Detection
4.12. Workflow for Oxygen Consumption, ATP and Lactate Assays
4.13. Oxygen Consumption Assay
4.14. ATP Assay
4.15. Lactate Assay
4.16. Western Blotting
4.17. Proteasome Activity
4.18. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RTT | Rett syndrome |
| MECP2 | Methyl-CpG binding Protein 2 |
| MSCs | Mesenchymal Stromal Cells |
| WT | Wild Type |
| CKIs | Cyclin Kinase Inhibitors |
| DDR | DNA Damage Response |
| ATM | Ataxia Telangiectasia Mutated kinase |
| 2-DG | 2-deoxy-d-Glucose |
| BPTES | Bis-2-(5-Phenylacetamido-1,3,4-Thiadiazol-2-yl)Ethyl Sulfide |
| FCCP | Trifluoromethoxy Carbonylcyanide Phenylhydrazone |
| LDH | Lactate Dehydrogenase |
| LC3 | Microtubule associated protein 1 Light Chain 3 |
| TCA | Tricarboxylic Acid |
| α-MEM | Minimal Essential Media with alpha modifications |
| FBS | Fetal Bovine Serum |
| bFGF | basic Fibroblast Growth Factor |
| PBS | Phosphate Buffered Saline |
| 4-MUG | 4-Methylumbelliferyl-β-d-Galactopyranoside |
| RT-qPCR | quantitative Reverse Transcription Polymerase Chain Reaction |
| DAPI | 4′,6-Diamidino-2-Phenylindole |
| OCR | Oxygen Consumption Rates |
| ETC | Electron Transport Chain |
| AMC | 7-Amino-4-methylcoumarin |
| mTOR | mechanistic Target of Rapamycin |
References
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Squillaro, T.; Alessio, N.; Cipollaro, M.; Renieri, A.; Giordano, A.; Galderisi, U. Partial silencing of methyl cytosine protein binding 2 (MECP2) in mesenchymal stem cells induces senescence with an increase in damaged DNA. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; Lasalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Luikenhuis, S.; Giacometti, E.; Beard, C.F.; Jaenisch, R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Tudor, M.; Akbarian, S.; Chen, R.Z.; Jaenisch, R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15536–15541. [Google Scholar] [CrossRef]
- Squillaro, T.; Alessio, N.; Cipollaro, M.; Melone, M.A.; Hayek, G.; Renieri, A.; Giordano, A.; Galderisi, U. Reduced expression of MECP2 affects cell commitment and maintenance in neurons by triggering senescence: New perspective for Rett syndrome. Mol. Biol. Cell 2012, 23, 1435–1445. [Google Scholar] [CrossRef]
- Squillaro, T.; Hayek, G.; Farina, E.; Cipollaro, M.; Renieri, A.; Galderisi, U. A case report: Bone marrow mesenchymal stem cells from a Rett syndrome patient are prone to senescence and show a lower degree of apoptosis. J. Cell. Biochem. 2008, 103, 1877–1885. [Google Scholar] [CrossRef]
- Alessio, N.; Riccitiello, F.; Squillaro, T.; Capasso, S.; Del Gaudio, S.; Di Bernardo, G.; Cipollaro, M.; Melone, M.A.B.; Peluso, G.; Galderisi, U. Neural stem cells from a mouse model of Rett syndrome are prone to senescence, show reduced capacity to cope with genotoxic stress, and are impaired in the differentiation process. Exp. Mol. Med. 2018, 50, 1. [Google Scholar] [CrossRef]
- Della Ragione, F.; Vacca, M.; Fioriniello, S.; Pepe, G.; D’Esposito, M. MECP2, a multi-talented modulator of chromatin architecture. Brief. Funct. Genomics 2016, 15, 420–431. [Google Scholar] [CrossRef]
- Ozkul, Y.; Galderisi, U. The Impact of Epigenetics on Mesenchymal Stem Cell Biology. J. Cell. Physiol. 2016, 231, 2393–2401. [Google Scholar] [CrossRef]
- Di Bernardo, G.; Cipollaro, M.; Galderisi, U. Chromatin modification and senescence. Curr. Pharm. Des. 2012, 18, 1686–1693. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Ng, H.H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef]
- Nacarelli, T.; Sell, C. Targeting metabolism in cellular senescence, a role for intervention. Mol. Cell. Endocrinol. 2017, 455, 83–92. [Google Scholar] [CrossRef]
- Capasso, S.; Alessio, N.; Squillaro, T.; Di Bernardo, G.; Melone, M.A.; Cipollaro, M.; Peluso, G.; Galderisi, U. Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget 2015, 6, 39457–39468. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy and senescence: A partnership in search of definition. Autophagy 2013, 9, 808–812. [Google Scholar] [CrossRef]
- White, E.; Lowe, S.W. Eating to exit: Autophagy-enabled senescence revealed. Genes Dev. 2009, 23, 784–787. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Stratford, F.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central role of the proteasome in senescence and survival of human fibroblasts: Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003, 278, 28026–28037. [Google Scholar] [CrossRef]
- Galderisi, U.; Giordano, A. The gap between the physiological and therapeutic roles of mesenchymal stem cells. Med. Res. Rev. 2014, 34, 1100–1126. [Google Scholar] [CrossRef]
- Alessio, N.; Capasso, S.; Di Bernardo, G.; Cappabianca, S.; Casale, F.; Calarco, A.; Cipollaro, M.; Peluso, G.; Galderisi, U. Mesenchymal stromal cells having inactivated RB1 survive following low irradiation and accumulate damaged DNA: Hints for side effects following radiotherapy. Cell Cycle 2017, 16, 251–258. [Google Scholar] [CrossRef]
- Alessio, N.; Squillaro, T.; Ozcan, S.; Di Bernardo, G.; Venditti, M.; Melone, M.; Peluso, G.; Galderisi, U. Stress and stem cells: Adult Muse cells tolerate extensive genotoxic stimuli better than mesenchymal stromal cells. Oncotarget 2018, 9, 19328–19341. [Google Scholar] [CrossRef]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials With Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef]
- Jori, F.P.; Napolitano, M.A.; Melone, M.A.; Cipollaro, M.; Cascino, A.; Giordano, A.; Galderisi, U. Role of RB and RB2/P130 genes in marrow stromal stem cells plasticity. J. Cell. Physiol. 2004, 200, 201–212. [Google Scholar] [CrossRef]
- Jefferson, A.; Leonard, H.; Siafarikas, A.; Woodhead, H.; Fyfe, S.; Ward, L.M.; Munns, C.; Motil, K.; Tarquinio, D.; Shapiro, J.R.; et al. Clinical Guidelines for Management of Bone Health in Rett Syndrome Based on Expert Consensus and Available Evidence. PLoS ONE 2016, 11, e0146824. [Google Scholar] [CrossRef]
- Galderisi, U.; Helmbold, H.; Squillaro, T.; Alessio, N.; Komm, N.; Khadang, B.; Cipollaro, M.; Bohn, W.; Giordano, A. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 2009, 18, 1033–1042. [Google Scholar] [CrossRef]
- Squillaro, T.; Antonucci, I.; Alessio, N.; Esposito, A.; Cipollaro, M.; Melone, M.A.B.; Peluso, G.; Stuppia, L.; Galderisi, U. Impact of lysosomal storage disorders on biology of mesenchymal stem cells: Evidences from in vitro silencing of glucocerebrosidase (GBA) and alpha-galactosidase A (GLA) enzymes. J. Cell. Physiol. 2017, 232, 3454–3467. [Google Scholar] [CrossRef]
- Di Bernardo, G.; Squillaro, T.; Dell’Aversana, C.; Miceli, M.; Cipollaro, M.; Cascino, A.; Altucci, L.; Galderisi, U. Histone deacetylase inhibitors promote apoptosis and senescence in human mesenchymal stem cells. Stem Cells Dev. 2009, 18, 573–581. [Google Scholar] [CrossRef]
- Alessio, N.; Del Gaudio, S.; Capasso, S.; Di Bernardo, G.; Cappabianca, S.; Cipollaro, M.; Peluso, G.; Galderisi, U. Low dose radiation induced senescence of human mesenchymal stromal cells and impaired the autophagy process. Oncotarget 2015, 6, 8155–8166. [Google Scholar] [CrossRef]
- Squillaro, T.; Severino, V.; Alessio, N.; Farina, A.; Di Bernardo, G.; Cipollaro, M.; Peluso, G.; Chambery, A.; Galderisi, U. De-regulated expression of the BRG1 chromatin remodeling factor in bone marrow mesenchymal stromal cells induces senescence associated with the silencing of NANOG and changes in the levels of chromatin proteins. Cell Cycle 2015, 14, 1315–1326. [Google Scholar] [CrossRef]
- Capasso, S.; Alessio, N.; Di Bernardo, G.; Cipollaro, M.; Melone, M.A.; Peluso, G.; Giordano, A.; Galderisi, U. Silencing of RB1 and RB2/P130 during adipogenesis of bone marrow stromal cells results in dysregulated differentiation. Cell Cycle 2014, 13, 482–490. [Google Scholar] [CrossRef]
- Olivieri, F.; Albertini, M.C.; Orciani, M.; Ceka, A.; Cricca, M.; Procopio, A.D.; Bonafe, M. DNA damage response (DDR) and senescence: Shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d’Adda di Fagagna, F. Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 2014, 9, e110969. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Alessio, N.; Di Bernardo, G.; Ozcan, S.; Peluso, G.; Galderisi, U. Stem Cells and DNA Repair Capacity: Muse Stem Cells Are Among the Best Performers. Adv. Exp. Med. Biol. 2018, 1103, 103–113. [Google Scholar]
- Freeman, A.K.; Monteiro, A.N. Phosphatases in the cellular response to DNA damage. Cell Commun. Signal. CCS 2010, 8, 27. [Google Scholar] [CrossRef]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef]
- Fu, S.; Yang, Y.; Das, T.K.; Yen, Y.; Zhou, B.S.; Zhou, M.M.; Ohlmeyer, M.; Ko, E.C.; Cagan, R.; Rosenstein, B.S.; et al. gamma-H2AX kinetics as a novel approach to high content screening for small molecule radiosensitizers. PLoS ONE 2012, 7, e38465. [Google Scholar] [CrossRef]
- Kao, J.; Milano, M.T.; Javaheri, A.; Garofalo, M.C.; Chmura, S.J.; Weichselbaum, R.R.; Kron, S.J. gamma-H2AX as a therapeutic target for improving the efficacy of radiation therapy. Curr. Cancer Drug Targets 2006, 6, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef]
- Calcada, D.; Vianello, D.; Giampieri, E.; Sala, C.; Castellani, G.; de Graaf, A.; Kremer, B.; van Ommen, B.; Feskens, E.; Santoro, A.; et al. The role of low-grade inflammation and metabolic flexibility in aging and nutritional modulation thereof: A systems biology approach. Mech. Ageing Dev. 2014, 136–137, 138–147. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Fenteany, G.; Schreiber, S.L. Lactacystin, proteasome function, and cell fate. J. Biol. Chem. 1998, 273, 8545–8548. [Google Scholar] [CrossRef]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16(INK4a) Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. [Google Scholar] [CrossRef]
- Helmbold, H.; Galderisi, U.; Bohn, W. The switch from pRb/p105 to Rb2/p130 in DNA damage and cellular senescence. J. Cell. Physiol. 2012, 227, 508–513. [Google Scholar] [CrossRef]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef]
- Xu, Y.; Price, B.D. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle 2011, 10, 261–267. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; House, N.; Price, B.D. Patching Broken DNA: Nucleosome Dynamics and the Repair of DNA Breaks. J. Mol. Biol. 2016, 428 Pt B, 1846–1860. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef]
- Liebhaber, G.M.; Riemann, E.; Baumeister, F.A. Ketogenic diet in Rett syndrome. J. Child Neurol. 2003, 18, 74–75. [Google Scholar] [CrossRef]
- Giampietro, P.F.; Schowalter, D.B.; Merchant, S.; Campbell, L.R.; Swink, T.; Roa, B.B. Widened clinical spectrum of the Q128P MECP2 mutation in Rett syndrome. Child’s Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2006, 22, 320–324. [Google Scholar] [CrossRef]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial Dysfunction in the Pathogenesis of Rett Syndrome: Implications for Mitochondria-Targeted Therapies. Front. Cell. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef]
- Mucerino, S.; Di Salle, A.; Alessio, N.; Margarucci, S.; Nicolai, R.; Melone, M.A.; Galderisi, U.; Peluso, G. Alterations in the carnitine cycle in a mouse model of Rett syndrome. Sci. Rep. 2017, 7, 41824. [Google Scholar] [CrossRef]
- Lappalainen, R.; Riikonen, R.S. Elevated CSF lactate in the Rett syndrome: Cause or consequence? Brain Dev. 1994, 16, 399–401. [Google Scholar] [CrossRef]
- De Felice, C.; Signorini, C.; Durand, T.; Ciccoli, L.; Leoncini, S.; D’Esposito, M.; Filosa, S.; Oger, C.; Guy, A.; Bultel-Ponce, V.; et al. Partial rescue of Rett syndrome by omega-3 polyunsaturated fatty acids (PUFAs) oil. Genes Nutr. 2012, 7, 447–458. [Google Scholar] [CrossRef]
- Gold, W.A.; Williamson, S.L.; Kaur, S.; Hargreaves, I.P.; Land, J.M.; Pelka, G.J.; Tam, P.P.; Christodoulou, J. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): Implications for the disease phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef]
- Park, M.J.; Aja, S.; Li, Q.; Degano, A.L.; Penati, J.; Zhuo, J.; Roe, C.R.; Ronnett, G.V. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE 2014, 9, e109527. [Google Scholar] [CrossRef]
- Sitte, N.; Merker, K.; Von Zglinicki, T.; Grune, T.; Davies, K.J. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: Part I—Effects of proliferative senescence. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 2495–2502. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genomics 2014, 15, 38–51. [Google Scholar] [CrossRef]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef]
- Zoghbi, H.Y. Rett Syndrome and the Ongoing Legacy of Close Clinical Observation. Cell 2016, 167, 293–297. [Google Scholar] [CrossRef]
- Humphreys, P.; Barrowman, N. The Incidence and Evolution of Parkinsonian Rigidity in Rett Syndrome: A Pilot Study. Can. J. Neurol. Sci. Le J. Can. Sci. Neurol. 2016, 43, 567–573. [Google Scholar] [CrossRef]
- Venkateswaran, S.; McMillan, H.J.; Doja, A.; Humphreys, P. Adolescent onset cognitive regression and neuropsychiatric symptoms associated with the A140V MECP2 mutation. Dev. Med. Child Neurol. 2014, 56, 91–94. [Google Scholar] [CrossRef]
- Roze, E.; Cochen, V.; Sangla, S.; Bienvenu, T.; Roubergue, A.; Leu-Semenescu, S.; Vidaihet, M. Rett syndrome: An overlooked diagnosis in women with stereotypic hand movements, psychomotor retardation, Parkinsonism, and dystonia? Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 387–389. [Google Scholar] [CrossRef]
- Samaco, R.C.; McGraw, C.M.; Ward, C.S.; Sun, Y.; Neul, J.L.; Zoghbi, H.Y. Female Mecp2(+/−) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum. Mol. Genet. 2013, 22, 96–109. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Gary, R.K.; Kindell, S.M. Quantitative assay of senescence-associated beta-galactosidase activity in mammalian cell extracts. Anal. Biochem. 2005, 343, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, A.; Di Salle, A.; Petillo, O.; Melone, M.A.; Grimaldi, G.; Bellotti, A.; Torelli, G.; De’ Santi, M.S.; Cantatore, G.; Marinelli, A.; et al. High grade glioblastoma is associated with aberrant expression of ZFP57, a protein involved in gene imprinting, and of CPT1A and CPT1C that regulate fatty acid metabolism. Cancer Biol. Ther. 2014, 15, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.; Giuliano, M.; Squillaro, T.; Alessio, N.; Casale, F.; Mattioli, E.; Cipollaro, M.; Giordano, A.; Galderisi, U. Genes involved in regulation of stem cell properties: Studies on their expression in a small cohort of neuroblastoma patients. Cancer Biol. Ther. 2009, 8, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Investig. 2013, 123, 980–989. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 2016, 8, 231–244. [Google Scholar] [CrossRef]
- Squillaro, T.; Schettino, C.; Sampaolo, S.; Galderisi, U.; Di Iorio, G.; Giordano, A.; Melone, M.A.B. Adult-onset brain tumors and neurodegeneration: Are polyphenols protective? J. Cell. Physiol. 2018, 233, 3955–3967. [Google Scholar] [CrossRef]
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharmacol. 2018, 154, 303–317. [Google Scholar] [CrossRef]
- Finicelli, M.; Squillaro, T.; Di Cristo, F.; Di Salle, A.; Melone, M.A.B.; Galderisi, U.; Peluso, G. Metabolic syndrome, Mediterranean diet, and polyphenols: Evidence and perspectives. J. Cell. Physiol. 2019, 234, 5807–5826. [Google Scholar] [CrossRef]
- Martel, J.; Ojcius, D.M.; Ko, Y.F.; Chang, C.J.; Young, J.D. Antiaging effects of bioactive molecules isolated from plants and fungi. Med. Res. Rev. 2019. [Google Scholar] [CrossRef]
- Serino, A.; Salazar, G. Protective Role of Polyphenols against Vascular Inflammation, Aging and Cardiovascular Disease. Nutrients 2018, 11, 53. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Squillaro, T.; Alessio, N.; Capasso, S.; Di Bernardo, G.; Melone, M.A.B.; Peluso, G.; Galderisi, U. Senescence Phenomena and Metabolic Alteration in Mesenchymal Stromal Cells from a Mouse Model of Rett Syndrome. Int. J. Mol. Sci. 2019, 20, 2508. https://doi.org/10.3390/ijms20102508
Squillaro T, Alessio N, Capasso S, Di Bernardo G, Melone MAB, Peluso G, Galderisi U. Senescence Phenomena and Metabolic Alteration in Mesenchymal Stromal Cells from a Mouse Model of Rett Syndrome. International Journal of Molecular Sciences. 2019; 20(10):2508. https://doi.org/10.3390/ijms20102508
Chicago/Turabian StyleSquillaro, Tiziana, Nicola Alessio, Stefania Capasso, Giovanni Di Bernardo, Mariarosa Anna Beatrice Melone, Gianfranco Peluso, and Umberto Galderisi. 2019. "Senescence Phenomena and Metabolic Alteration in Mesenchymal Stromal Cells from a Mouse Model of Rett Syndrome" International Journal of Molecular Sciences 20, no. 10: 2508. https://doi.org/10.3390/ijms20102508
APA StyleSquillaro, T., Alessio, N., Capasso, S., Di Bernardo, G., Melone, M. A. B., Peluso, G., & Galderisi, U. (2019). Senescence Phenomena and Metabolic Alteration in Mesenchymal Stromal Cells from a Mouse Model of Rett Syndrome. International Journal of Molecular Sciences, 20(10), 2508. https://doi.org/10.3390/ijms20102508