Computational and Experimental Tools to Monitor the Changes in Translation Efficiency of Plant mRNA on a Genome-Wide Scale: Advantages, Limitations, and Solutions


Abstract
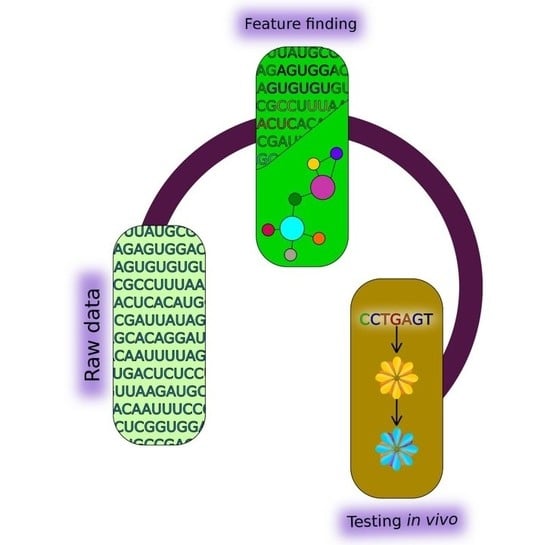
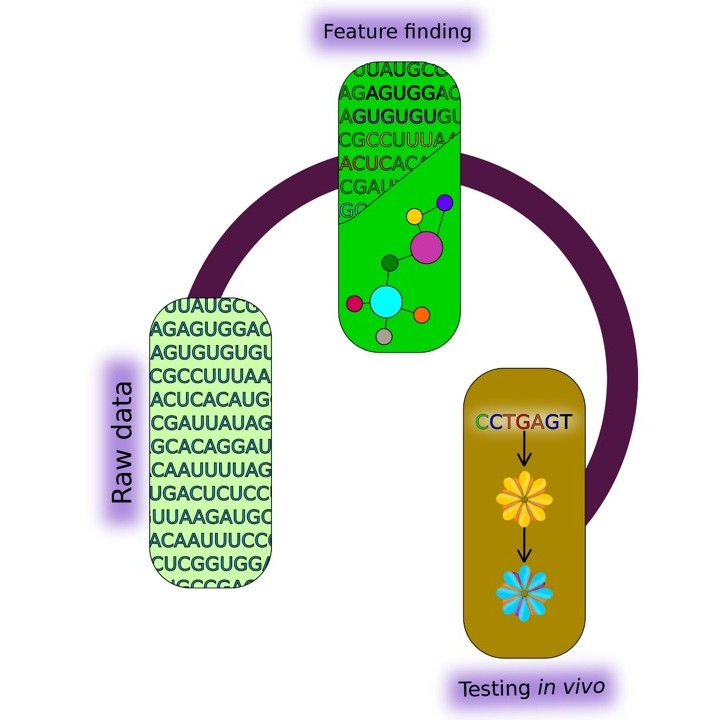
1. Introduction
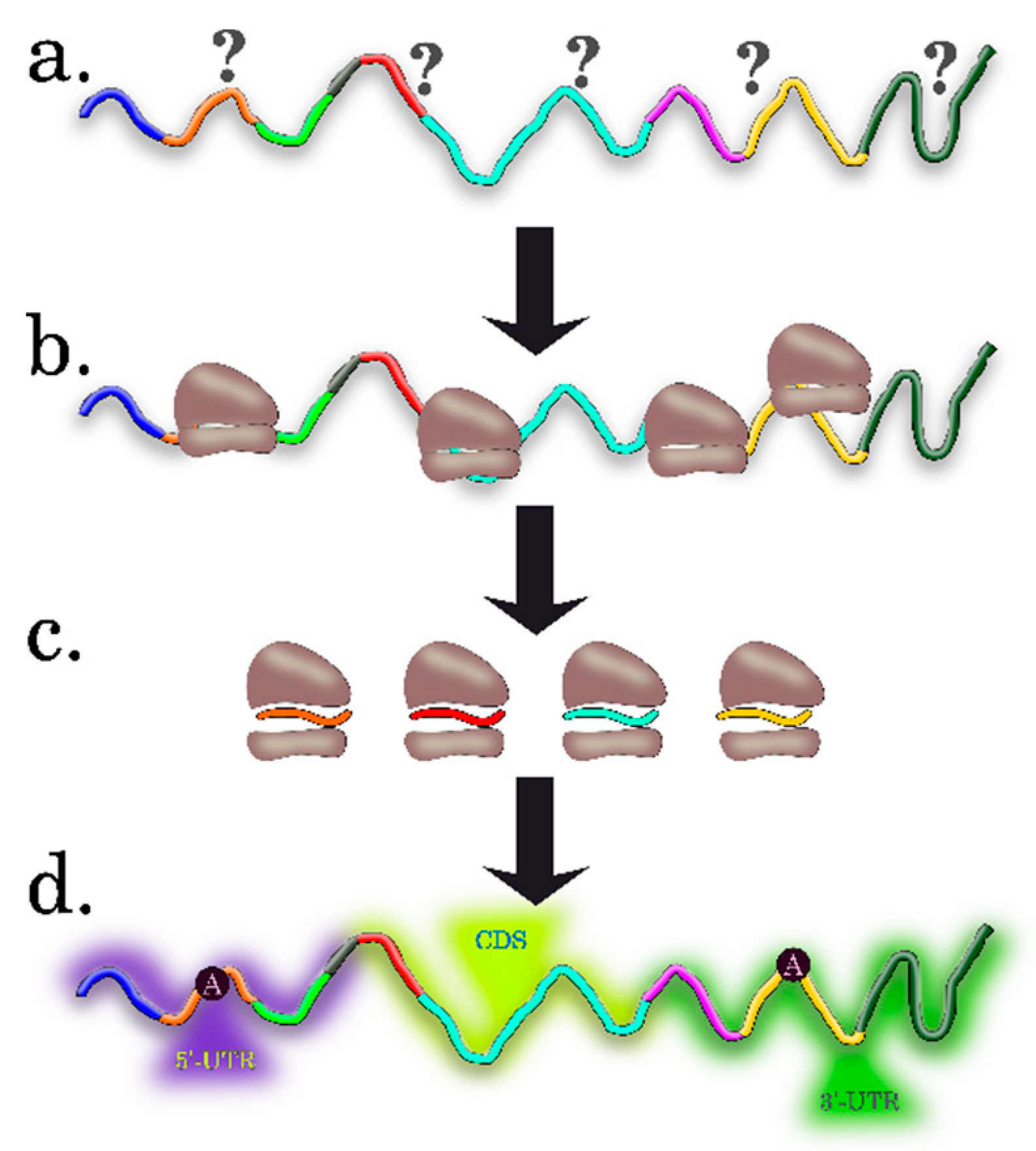
2. Experimental Approaches to Determine Differentially-Translated mRNAs in Plants
2.1. Profiling Polysomes
2.2. Translating Ribosome Affinity Purification (TRAP) and TRAP-Seq
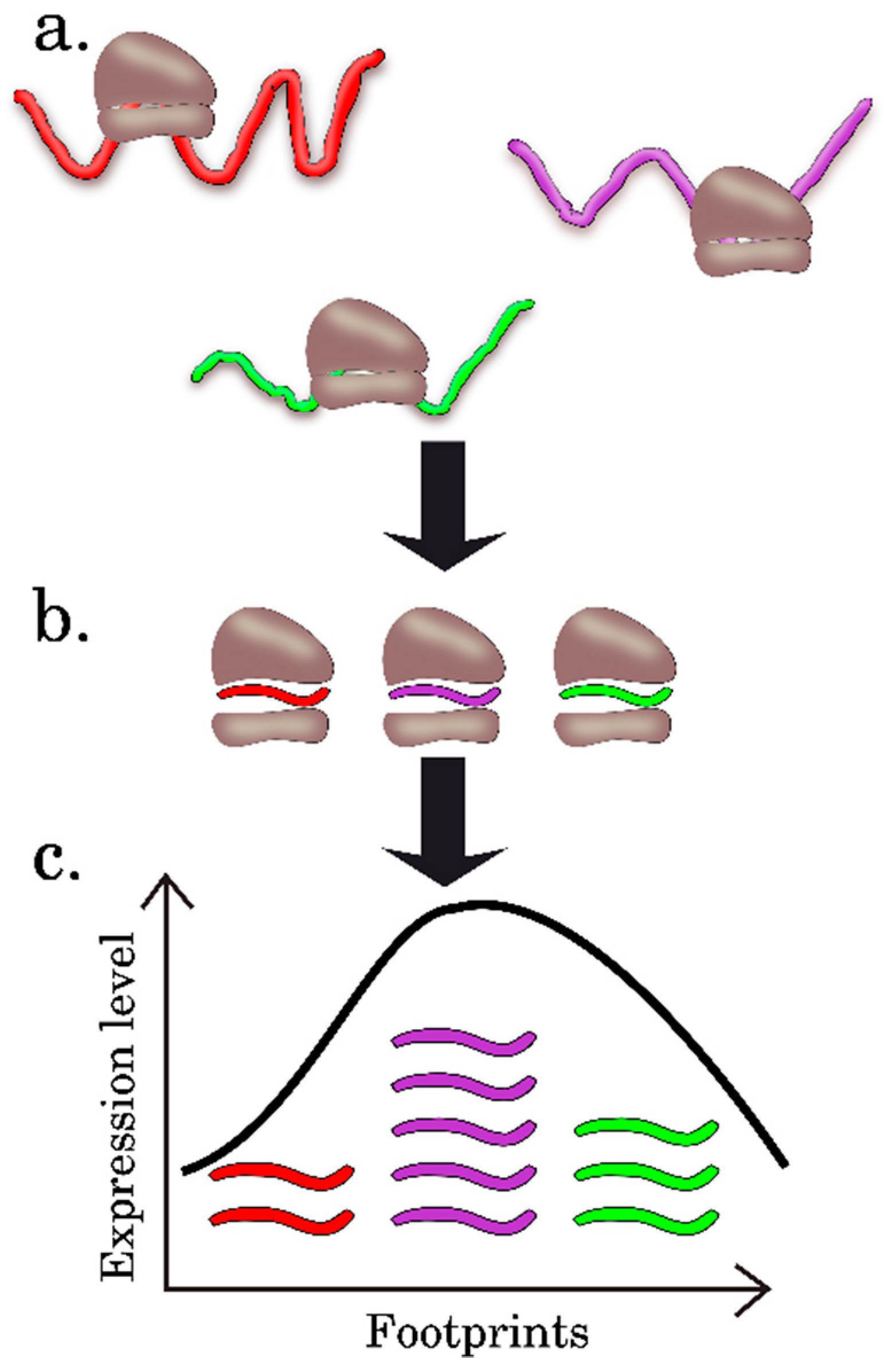
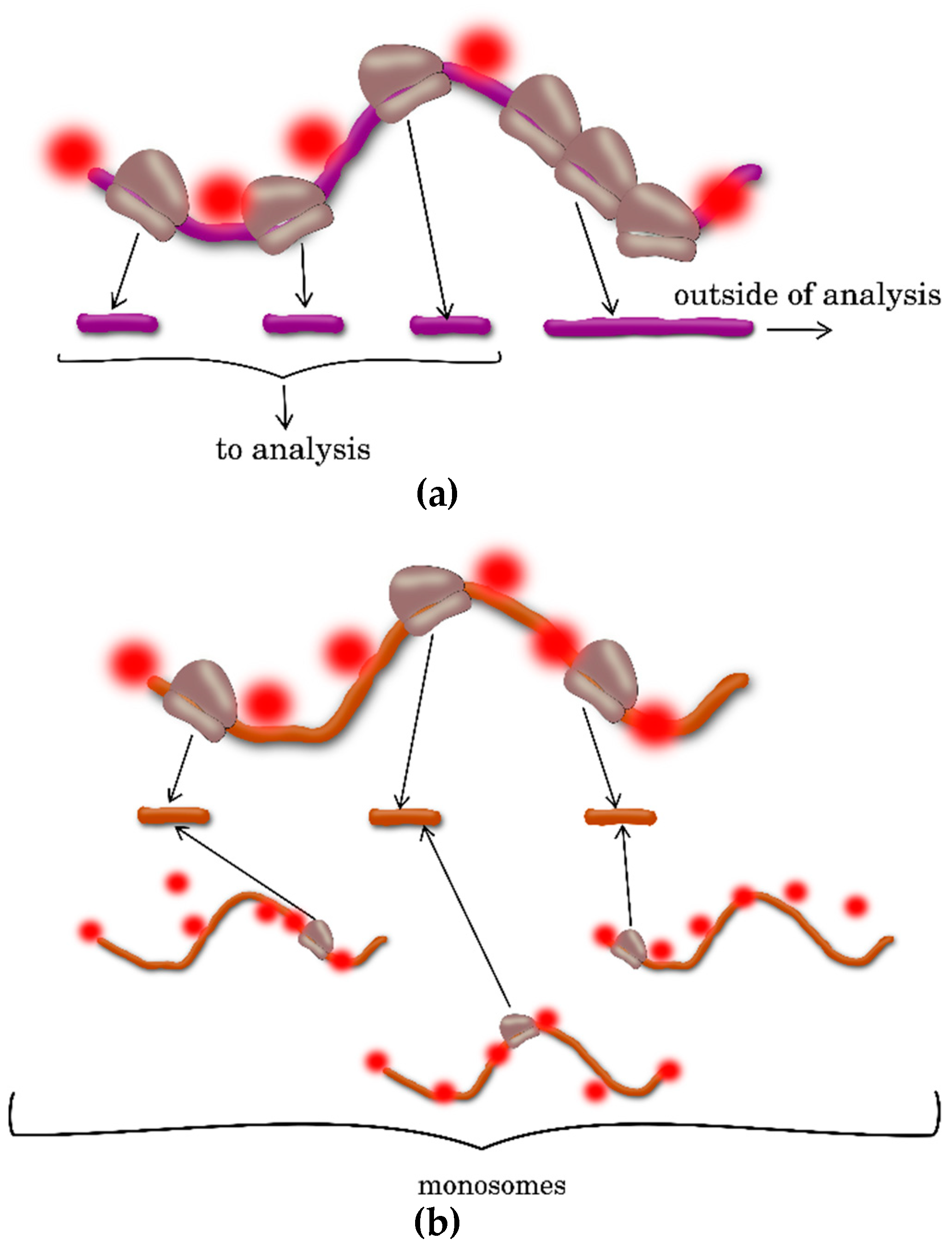
2.3. Ribosome Profiling, or Ribo-Seq
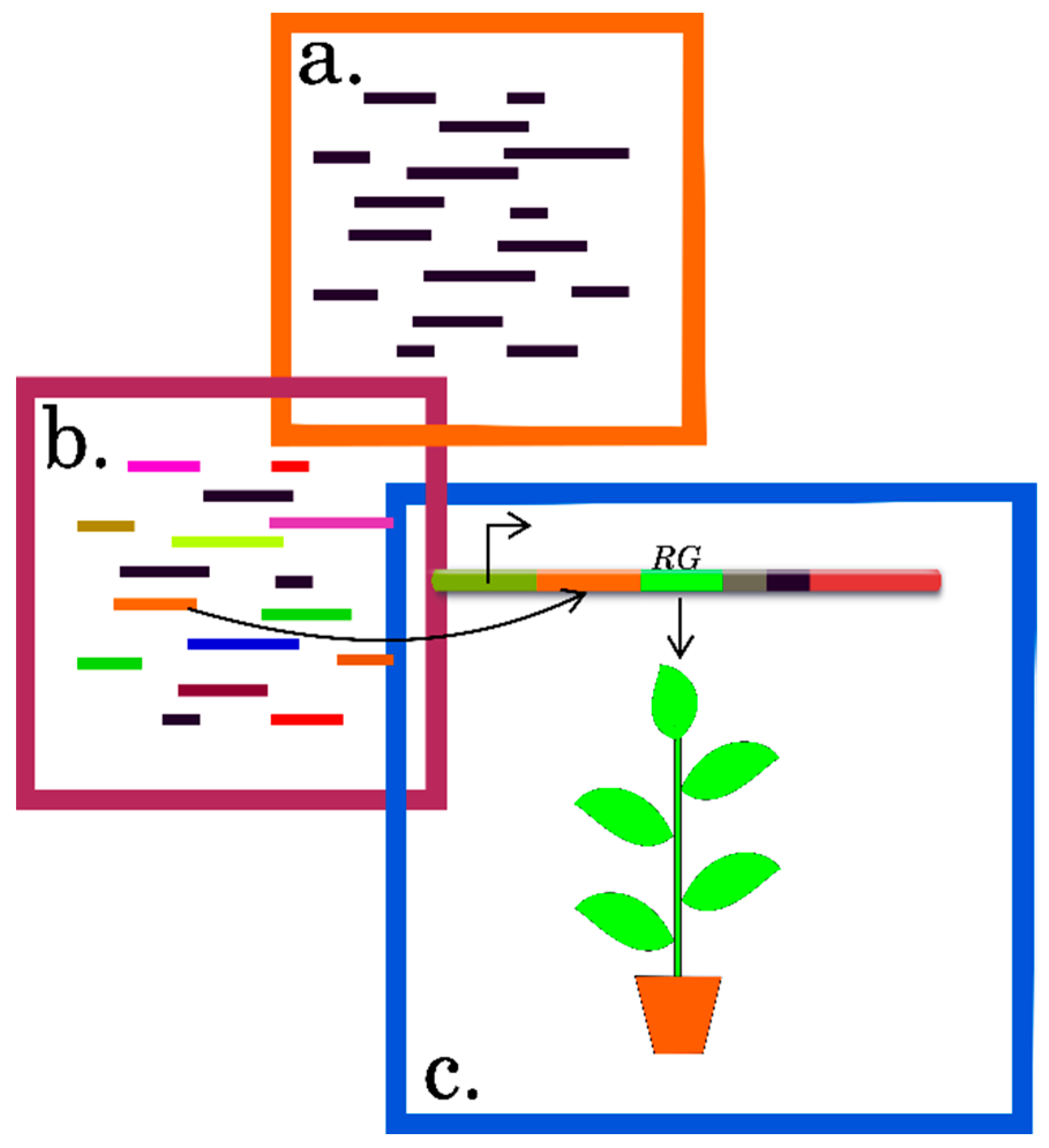
3. Computational Algorithms for Predicting the Features of Plant mRNAs Important for Differential Translation
3.1. Preparing Datasets for Analysis
3.2. Statistical Methods
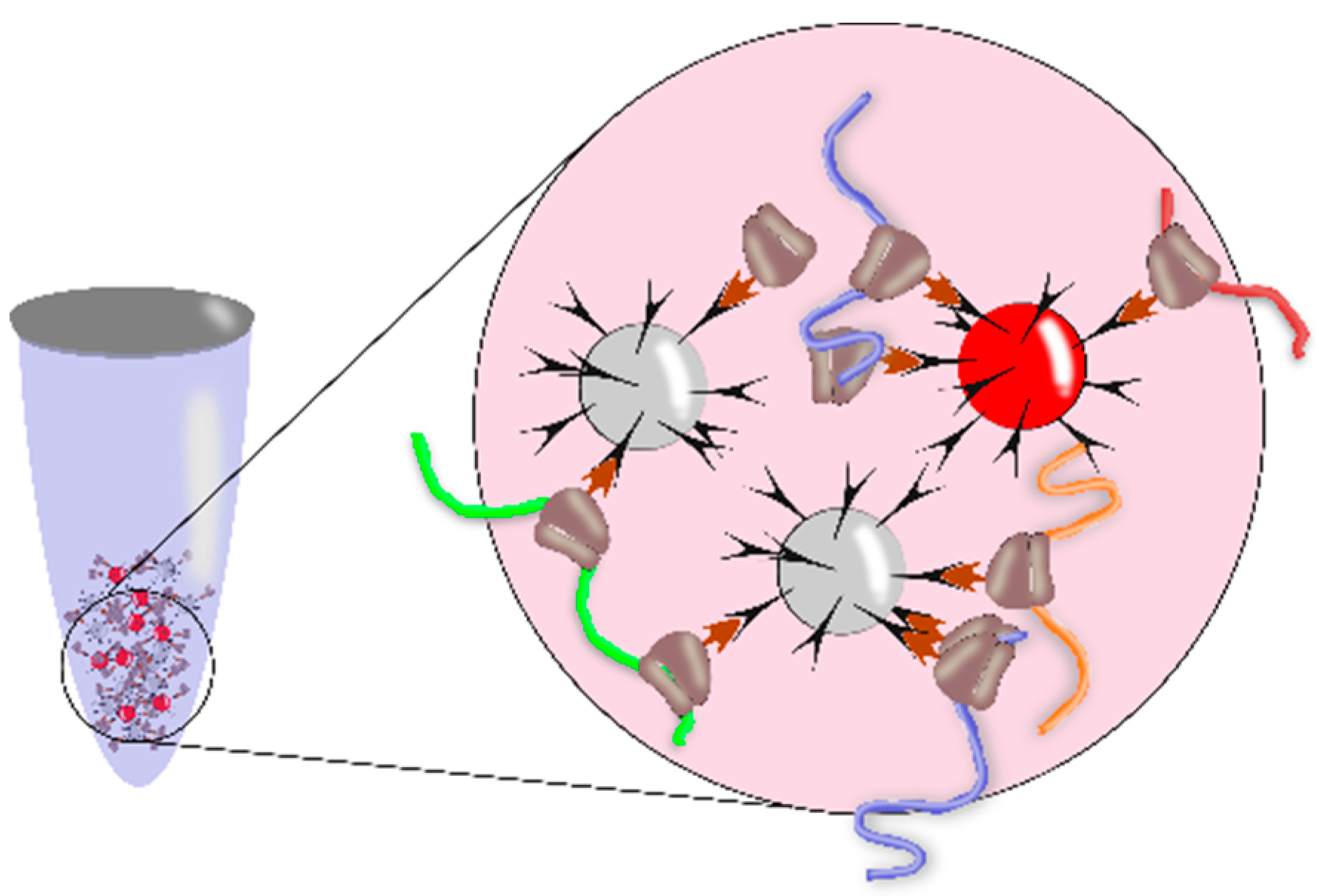
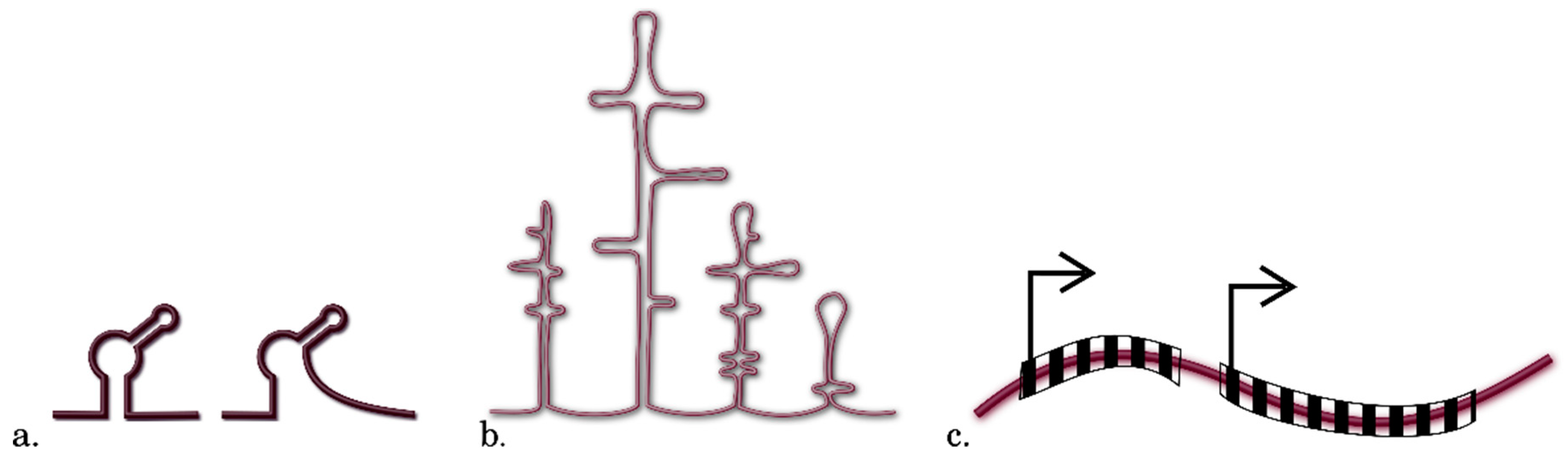
3.3. Methods for Identification of RNA Motifs
3.4. Detection of Other Context Features of RNA
4. Approaches for Experimental Verification of the Systemic Experimental Data and Theoretical Predictions of Intrinsic Features of Plant mRNAs Important for Differential Translation
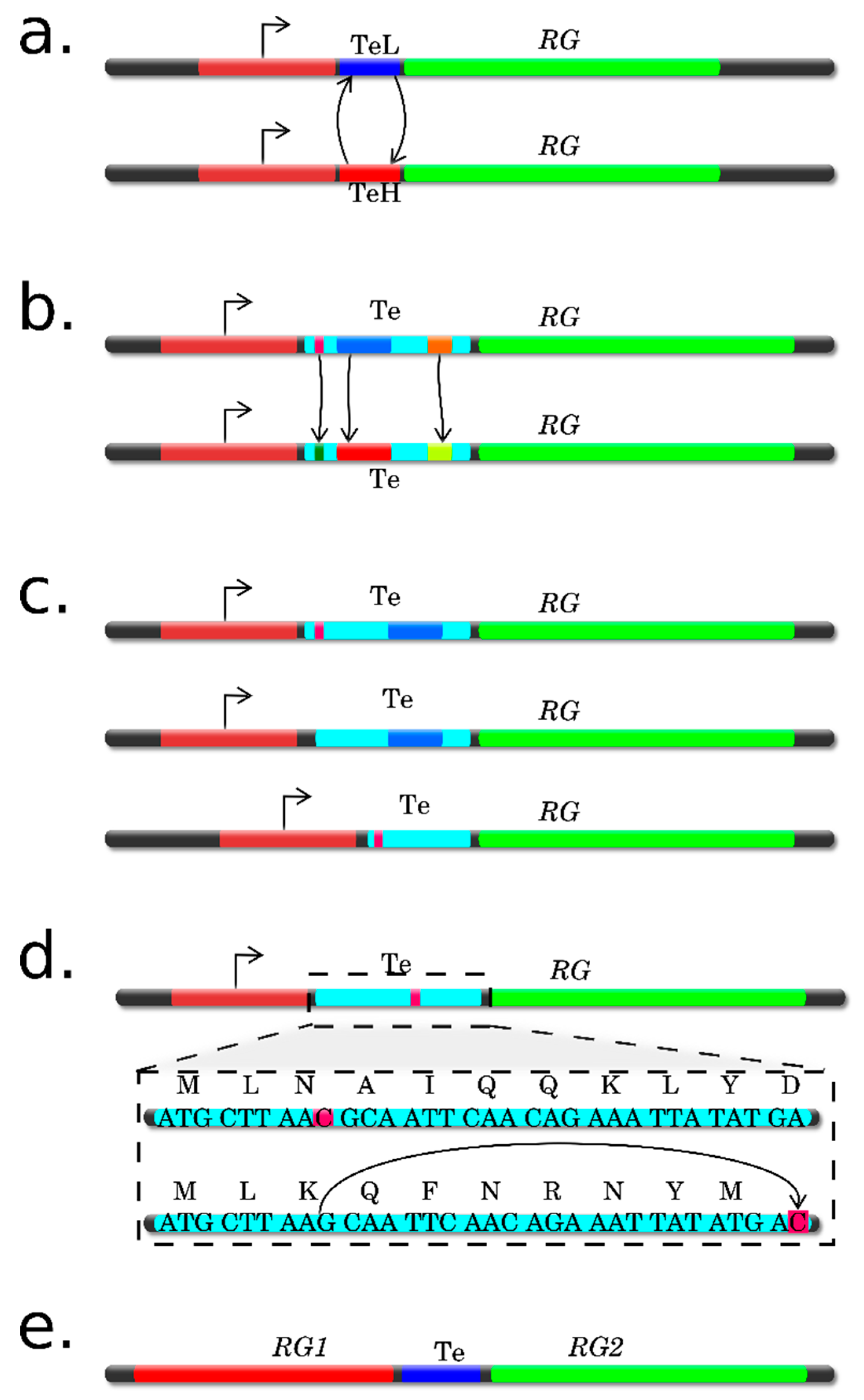
4.1. Direct Measurments of Individual Transcripts
4.2. Site-Specific Substitutions
4.3. Analysis Using Deletions
4.4. Translation from Alternative ORFs
4.5. Frameshift Mutations
4.6. Characteristics and Reasoning for Selection of a Verification Method(s)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Baerenfaller, K.; Grossmann, J.; Grobei, M.A.; Hull, R.; Hirsch-Hoffmann, M.; Yalovsky, S.; Zimmermann, P.; Grossniklaus, U.; Gruissem, W.; Baginsky, S. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 2008, 320, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Bailey-Serres, J. mRNA sequence features that contribute to translational regulation in Arabidopsis. Nucleic Acids Res. 2005, 33, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Sablok, G.; Powell, J.J.; Kazan, K. Emerging Roles and Landscape of Translating mRNAs in Plants. Front. Plant Sci. 2017, 8, 1443. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni-Putman, S.M.; Stepanova, A.N. A Plant Biologist’s Toolbox to Study Translation. Front. Plant Sci. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Lecampion, C.; Floris, M.; Fantino, J.R.; Robaglia, C.; Laloi, C. An Easy Method for Plant Polysome Profiling. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Salwan, R.; Sharma, P.N.; Gulati, A. Integrated Translatome and Proteome: Approach for Accurate Portraying of Widespread Multifunctional Aspects of Trichoderma. Front. Microbiol. 2017, 8, 1602. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Matsuura, H.; Demura, T.; Kato, K. Changes in Polysome Association of mRNA Throughout Growth and Development in Arabidopsis thaliana. Plant Cell Physiol. 2015, 56, 2169–2180. [Google Scholar] [CrossRef]
- Matsuura, H.; Takenami, S.; Kubo, Y.; Ueda, K.; Ueda, A.; Yamaguchi, M.; Hirata, K.; Demura, T.; Kanaya, S.; Kato, K. A computational and experimental approach reveals that the 5’-proximal region of the 5’-UTR has a Cis-regulatory signature responsible for heat stress-regulated mRNA translation in Arabidopsis. Plant Cell Physiol. 2013, 54, 474–483. [Google Scholar] [CrossRef]
- Yamasaki, S.; Sanada, Y.; Imase, R.; Matsuura, H.; Ueno, D.; Demura, T.; Kato, K. Arabidopsis thaliana cold-regulated 47 gene 5′-untranslated region enables stable high-level expression of transgenes. J. Biosci. Bioeng. 2018, 125, 124–130. [Google Scholar] [CrossRef]
- Matsuura, H.; Ishibashi, Y.; Shinmyo, A.; Kanaya, S.; Kato, K. Genome-wide analyses of early translational responses to elevated temperature and high salinity in Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 448–462. [Google Scholar] [CrossRef]
- Juntawong, P.; Hummel, M.; Bazin, J.; Bailey-Serres, J. Ribosome profiling: A tool for quantitative evaluation of dynamics in mRNA translation. Methods Mol. Biol. 2015, 1284, 139–173. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M.E.; Chang, I.F.; Gong, F.; Galbraith, D.W.; Bailey-Serres, J. Immunopurification of polyribosomal complexes of Arabidopsis for global analysis of gene expression. Plant Physiol. 2005, 138, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, A.; Juntawong, P.; Bailey-Serres, J. Isolation of plant polysomal mRNA by differential centrifugation and ribosome immunopurification methods. Methods Mol. Biol. 2009, 553, 109–126. [Google Scholar] [CrossRef]
- Mustroph, A.; Zanetti, M.E.; Girke, T.; Bailey-Serres, J. Isolation and analysis of mRNAs from specific cell types of plants by ribosome immunopurification. Methods Mol. Biol. 2013, 959, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chen, P.W.; Chuang, M.H.; Juntawong, P.; Bailey-Serres, J.; Jauh, G.Y. Profiling of translatomes of in vivo-grown pollen tubes reveals genes with roles in micropylar guidance during pollination in Arabidopsis. Plant Cell 2014, 26, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Meyerowitz, E.M. Cell-type specific analysis of translating RNAs in developing flowers reveals new levels of control. Mol. Syst. Biol. 2010, 6, 419. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.; Bailey-Serres, J. Selective mRNA sequestration by OLIGOURIDYLATE-BINDING PROTEIN 1 contributes to translational control during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- McGlincy, N.J.; Ingolia, N.T. Transcriptome-wide measurement of translation by ribosome profiling. Methods 2017, 126, 112–129. [Google Scholar] [CrossRef]
- Chotewutmontri, P.; Stiffler, N.; Watkins, K.P.; Barkan, A. Ribosome Profiling in Maize. Methods Mol. Biol. 2018, 1676, 165–183. [Google Scholar] [CrossRef]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–212. [Google Scholar] [CrossRef] [PubMed]
- Planchard, N.; Bertin, P.; Quadrado, M.; Dargel-Graffin, C.; Hatin, I.; Namy, O.; Mireau, H. The translational landscape of Arabidopsis mitochondria. Nucleic Acids Res. 2018, 46, 6218–6228. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, M.V.; Gladyshev, V.N. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 2017, 45, e6. [Google Scholar] [CrossRef] [PubMed]
- Eastman, G.; Smircich, P.; Sotelo-Silveira, J.R. Following Ribosome Footprints to Understand Translation at a Genome Wide Level. Comput. Struct. Biotechnol. J. 2018, 16, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Benfey, P.N. Small but Mighty: Functional Peptides Encoded by Small ORFs in Plants. Proteomics 2018, 18, e1700038. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Shi, J.; Chen, J.; Zhang, M.; Sun, S.; Xie, S.; Li, X.; Zeng, B.; Peng, L.; Hauck, A.; et al. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 2015, 84, 1206–1218. [Google Scholar] [CrossRef]
- Hsu, P.Y.; Calviello, L.; Wu, H.L.; Li, F.W.; Rothfels, C.J.; Ohler, U.; Benfey, P.N. Super-resolution ribosome profiling reveals unannotated translation events in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, E7126–E7135. [Google Scholar] [CrossRef]
- Lukoszek, R.; Feist, P.; Ignatova, Z. Insights into the adaptive response of Arabidopsis thaliana to prolonged thermal stress by ribosomal profiling and RNA-Seq. BMC Plant Biol. 2016, 16, 221. [Google Scholar] [CrossRef]
- Chotewutmontri, P.; Barkan, A. Multilevel effects of light on ribosome dynamics in chloroplasts program genome-wide and psbA-specific changes in translation. PLoS Genet. 2018, 14, e1007555. [Google Scholar] [CrossRef]
- Andreev, D.E.; O’Connor, P.B.; Loughran, G.; Dmitriev, S.E.; Baranov, P.V.; Shatsky, I.N. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2017, 45, 513–526. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–1210. [Google Scholar] [CrossRef]
- Yanguez, E.; Castro-Sanz, A.B.; Fernandez-Bautista, N.; Oliveros, J.C.; Castellano, M.M. Analysis of genome-wide changes in the translatome of Arabidopsis seedlings subjected to heat stress. PLoS ONE 2013, 8, e71425. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Wu, S.H.; Chen, H.M.; Wu, S.H. Widespread translational control contributes to the regulation of Arabidopsis photomorphogenesis. Mol. Syst. Biol. 2012, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Basbouss-Serhal, I.; Pateyron, S.; Cochet, F.; Leymarie, J.; Bailly, C. 5′ to 3′ mRNA Decay Contributes to the Regulation of Arabidopsis Seed Germination by Dormancy. Plant Physiol. 2017, 173, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Mustafaev, O.; Sadovskaya, N.S.; Tyurin, A.A.; Goldenkova-Pavlova, I.V. JetGene: An integrated database for analysis of regulatory regions or nucleotide contexts in plant differentially translated transcripts. manuscript in preparation.
- Deyneko, I.V.; Kel, A.E.; Bloecker, H.; Kauer, G. Signal-theoretical DNA similarity measure revealing unexpected similarities of E. coli promoters. In Silico Biol. 2005, 5, 547–555. [Google Scholar] [PubMed]
- Deyneko, I.V.; Kalybaeva, Y.M.; Kel, A.E.; Blöcker, H. Human-chimpanzee promoter comparisons: Property-conserved evolution? Genomics 2010, 96, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–49. [Google Scholar] [CrossRef]
- Williams, B.P.; Burgess, S.J.; Reyna-Llorens, I.; Knerova, J.; Aubry, S.; Stanley, S.; Hibberd, J.M. An Untranslated cis-Element Regulates the Accumulation of Multiple C4 Enzymes in Gynandropsis gynandra Mesophyll Cells. Plant Cell 2016, 28, 454–465. [Google Scholar] [CrossRef]
- Merchante, C.; Stepanova, A.N.; Alonso, J.M. Translation regulation in plants: An interesting past, an exciting present and a promising future. Plant J. 2017, 90, 628–653. [Google Scholar] [CrossRef]
- Thompson, S.R. So you want to know if your message has an IRES? Wiley Interdiscip. Rev. RNA 2012, 3, 697–705. [Google Scholar] [CrossRef]
- Cobbold, L.C.; Spriggs, K.A.; Haines, S.J.; Dobbyn, H.C.; Hayes, C.; de Moor, C.H.; Lilley, K.S.; Bushell, M.; Willis, A.E. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol. Cell. Biol. 2008, 28, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Sheshukova, E.V.; Komarova, T.V.; Ershova, N.M.; Shindyapina, A.V.; Dorokhov, Y.L. An Alternative Nested Reading Frame May Participate in the Stress-Dependent Expression of a Plant Gene. Front. Plant Sci. 2017, 8, 2137. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Rothnagel, J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Takahashi, A.; Naito, S.; Onouchi, H. BAIUCAS: A novel BLAST-based algorithm for the identification of upstream open reading frames with conserved amino acid sequences and its application to the Arabidopsis thaliana genome. Bioinformatics 2012, 28, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Ebina, I.; Takemoto-Tsutsumi, M.; Watanabe, S.; Koyama, H.; Endo, Y.; Kimata, K.; Igarashi, T.; Murakami, K.; Kudo, R.; Ohsumi, A.; et al. Identification of novel Arabidopsis thaliana upstream open reading frames that control expression of the main coding sequences in a peptide sequence-dependent manner. Nucleic Acids Res. 2015, 43, 1562–1576. [Google Scholar] [CrossRef] [PubMed]
- Tunney, R.; McGlincy, N.J.; Graham, M.E.; Naddaf, N.; Pachter, L.; Lareau, L.F. Accurate design of translational output by a neural network model of ribosome distribution. Nat. Struct. Mol. Biol. 2018, 25, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.T.; Kuintzle, R.; Teegarden, A.; Merrill, E., 3rd; Danaee, P.; Hendrix, D.A. A deep recurrent neural network discovers complex biological rules to decipher RNA protein-coding potential. Nucleic Acids Res. 2018, 46, 8105–8113. [Google Scholar] [CrossRef] [PubMed]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-nnet: An artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, S. Simple tricks of convolutional neural network architectures improve DNA-protein binding prediction. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Tyurin, A.A.; Kabardaeva, K.V.; Gra, O.A.; Mustafaev, O.M.; Sadovskaya, N.S.; Pavlenko, O.S.; Goldenkova-Pavlov, I.V. Efficient expression of a heterologous gene in plants depends on the nucleotide composition of mRNA’s 5’-region. Russ. J. Plant. Physiol. 2016, 4, 511–522. [Google Scholar] [CrossRef]
- Anami, S.; Njuguna, E.; Coussens, G.; Aesaert, S.; Van Lijsebettens, M. Higher plant transformation: Principles and molecular tools. Int. J. Dev. Biol. 2013, 57, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Cho, H.S.; Nam, H.; Jo, H.; Yoon, J.; Park, C.; Dang, T.V.T.; Kim, E.; Jeong, J.; Park, S.; et al. Translational control of phloem development by RNA G-quadruplex-JULGI determines plant sink strength. Nat. Plants 2018, 4, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.; Voss, B.; Maass, D.; Wust, F.; Schaub, P.; Beyer, P.; Welsch, R. Carotenogenesis Is Regulated by 5’UTR-Mediated Translation of Phytoene Synthase Splice Variants. Plant Physiol. 2016, 172, 2314–2326. [Google Scholar] [CrossRef]
- Dorokhov, Y.L.; Skulachev, M.V.; Ivanov, P.A.; Zvereva, S.D.; Tjulkina, L.G.; Merits, A.; Gleba, Y.Y.; Hohn, T.; Atabekov, J.G. Polypurine (A)-rich sequences promote cross-kingdom conservation of internal ribosome entry. Proc. Natl. Acad. Sci. USA 2002, 99, 5301–5306. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Schumacher, H.M.; Heine-Dobbernack, E.; El-Banna, A.; Hafeez, F.Y.; Jacobsen, H.J.; Kiesecker, H. Dicistronic binary vector system-A versatile tool for gene expression studies in cell cultures and plants. J. Biotechnol. 2010, 145, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gonzalez, A.S.; Fernandez, N.; Martinez-Salas, E.; Sanchez de Jimenez, E. Functional and structural analysis of maize hsp101 IRES. PLoS ONE 2014, 9, e107459. [Google Scholar] [CrossRef]
- Cui, Y.; Rao, S.; Chang, B.; Wang, X.; Zhang, K.; Hou, X.; Zhu, X.; Wu, H.; Tian, Z.; Zhao, Z.; et al. AtLa1 protein initiates IRES-dependent translation of WUSCHEL mRNA and regulates the stem cell homeostasis of Arabidopsis in response to environmental hazards. Plant Cell Environ. 2015, 38, 2098–2114. [Google Scholar] [CrossRef]
- Jorgensen, R.A.; Dorantes-Acosta, A.E. Conserved Peptide Upstream Open Reading Frames are Associated with Regulatory Genes in Angiosperms. Front. Plant Sci. 2012, 3, 191. [Google Scholar] [CrossRef]
- Tanaka, M.; Sotta, N.; Yamazumi, Y.; Yamashita, Y.; Miwa, K.; Murota, K.; Chiba, Y.; Hirai, M.Y.; Akiyama, T.; Onouchi, H.; et al. The Minimum Open Reading Frame, AUG-Stop, Induces Boron-Dependent Ribosome Stalling and mRNA Degradation. Plant Cell 2016, 28, 2830–2849. [Google Scholar] [CrossRef]
- Xu, G.; Yuan, M.; Ai, C.; Liu, L.; Zhuang, E.; Karapetyan, S.; Wang, S.; Dong, X. uORF-mediated translation allows engineered plant disease resistance without fitness costs. Nature 2017, 545, 491–494. [Google Scholar] [CrossRef]
- Hayashi, N.; Sasaki, S.; Takahashi, H.; Yamashita, Y.; Naito, S.; Onouchi, H. Identification of Arabidopsis thaliana upstream open reading frames encoding peptide sequences that cause ribosomal arrest. Nucleic Acids Res. 2017, 45, 8844–8858. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Elledge, S.J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 2007, 4, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Smedley, M.A.; Harwood, W.A. Gateway(R)-compatible plant transformation vectors. Methods Mol. Biol. 2015, 1223, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Tsvetanova, B.; Peng, L.; Liang, X.; Li, K.; Yang, J.P.; Ho, T.; Shirley, J.; Xu, L.; Potter, J.; Kudlicki, W.; et al. Genetic assembly tools for synthetic biology. Methods Enzymol. 2011, 498, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden gate shuffling: A one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Kandzia, R.; Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Tian, J. Circular polymerase extension cloning. Methods Mol. Biol. 2014, 1116, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Marillonnet, S. Golden Gate cloning. Methods Mol. Biol. 2014, 1116, 119–131. [Google Scholar] [CrossRef]
- Sarrion-Perdigones, A.; Vazquez-Vilar, M.; Palaci, J.; Castelijns, B.; Forment, J.; Ziarsolo, P.; Blanca, J.; Granell, A.; Orzaez, D. GoldenBraid 2.0: A comprehensive DNA assembly framework for plant synthetic biology. Plant Physiol. 2013, 162, 1618–1631. [Google Scholar] [CrossRef]
- Vyacheslavova, A.O.; Berdichevets, I.N.; Tyurin, A.A.; Shimshilashvili, K.R.; Mustafaev, O.N.; Goldenkova-Pavlova, I.V. Expression of heterologous genes in plant systems: New possibilities. Russ. J. Genet. 2012, 48, 1067–1079. [Google Scholar] [CrossRef]
- Agarwal, P.; Garg, V.; Gautam, T.; Pillai, B.; Kanoria, S.; Burma, P.K. A study on the influence of different promoter and 5’UTR (URM) cassettes from Arabidopsis thaliana on the expression level of the reporter gene beta glucuronidase in tobacco and cotton. Transgenic Res. 2014, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Tyurin, A.A.; Kabardaeva, K.V.; Berestovoy, M.A.; Sidorchuk, Y.V.; Fomenkov, A.A.; Nosov, A.V.; Goldenkova-Pavlova, I.V. Simple and reliable system for transient gene expression for the characteristic signal sequences and the estimation of the localization of target protein in plant cell. Russ. J. Plant. Physiol. 2017, 64, 672–679. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Approach | Basic Protocol | Advantages | Limitations | References |
|---|---|---|---|---|
| Polysome Profiling | Separation of transcripts with different ribosome loading by ultracentrifugation; supplemented by sequencing of different mRNA fractions, including transcriptome-wide analysis | Simplicity and possibility to analyze the plant species with annotated and unannotated genomes | Does not assess the number and location of ribosomes on each transcript | [4,5,21] |
| Translating Ribosome Affinity Purification | Separation of the transcripts with different ribosome loadings by absorption on anti-FLAG-M2 agarose; supplemented by mRNA sequencing, including transcriptome-wide analysis | Profiling of actively-translated RNAs from different plant tissues and particular cell types; Identification of only the mRNAs bound to ribosomes, which makes it possible to avoid the potential confusion with the transcripts associated with the other RNA-binding proteins | Cannot estimate the number and location of ribosomes on each transcript; Requires production and accurate selection of the plant transgenic lines that express an epitope-tagged variant of ribosomal protein L18 | [3,12,14,15] |
| Ribosome Profiling | Isolation and sequencing of the ribosome-protected mRNA fragments; modification of the protocol is necessary for individual species | Identification of the ribosome number and location on each transcript; Detection of new translated ORFs and noncanonical translation start sites | Requires significant material, time and labor investments; A considerable amount of biological material is necessary; The transcript regions with stacked ribosomes may be underrepresented; Incorrect trace identification may result from the RNA interaction with RNA-binding proteins of an analogous size; Applicable only to the plants with a well-annotated genome | [4,18,19,20,24] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldenkova-Pavlova, I.V.; Pavlenko, O.S.; Mustafaev, O.N.; Deyneko, I.V.; Kabardaeva, K.V.; Tyurin, A.A. Computational and Experimental Tools to Monitor the Changes in Translation Efficiency of Plant mRNA on a Genome-Wide Scale: Advantages, Limitations, and Solutions. Int. J. Mol. Sci. 2019, 20, 33. https://doi.org/10.3390/ijms20010033
Goldenkova-Pavlova IV, Pavlenko OS, Mustafaev ON, Deyneko IV, Kabardaeva KV, Tyurin AA. Computational and Experimental Tools to Monitor the Changes in Translation Efficiency of Plant mRNA on a Genome-Wide Scale: Advantages, Limitations, and Solutions. International Journal of Molecular Sciences. 2019; 20(1):33. https://doi.org/10.3390/ijms20010033
Chicago/Turabian StyleGoldenkova-Pavlova, Irina V., Olga S. Pavlenko, Orkhan N. Mustafaev, Igor V. Deyneko, Ksenya V. Kabardaeva, and Alexander A. Tyurin. 2019. "Computational and Experimental Tools to Monitor the Changes in Translation Efficiency of Plant mRNA on a Genome-Wide Scale: Advantages, Limitations, and Solutions" International Journal of Molecular Sciences 20, no. 1: 33. https://doi.org/10.3390/ijms20010033
APA StyleGoldenkova-Pavlova, I. V., Pavlenko, O. S., Mustafaev, O. N., Deyneko, I. V., Kabardaeva, K. V., & Tyurin, A. A. (2019). Computational and Experimental Tools to Monitor the Changes in Translation Efficiency of Plant mRNA on a Genome-Wide Scale: Advantages, Limitations, and Solutions. International Journal of Molecular Sciences, 20(1), 33. https://doi.org/10.3390/ijms20010033