Vitamin D Supplementation in Central Nervous System Demyelinating Disease—Enough Is Enough

{kind=link}
Abstract
1. Introduction
2. Vitamin D and Multiple Sclerosis
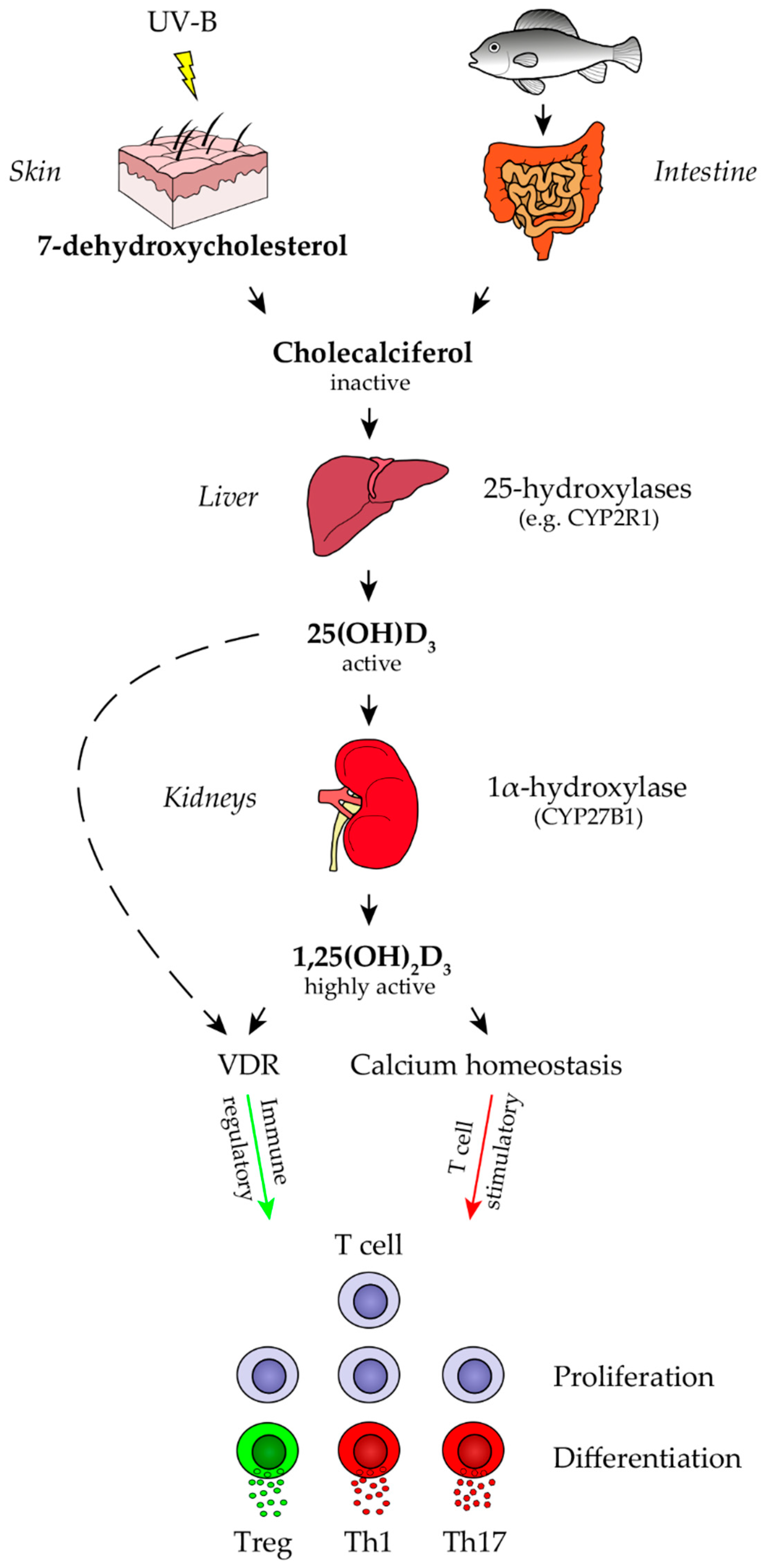
2.1. Vitamin D Metabolism and Direct Effects of Vitamin D Metabolites
2.2. Vitamin D Supplementation Studies in MS
2.3. Possible Side Effects of Secondary Hypercalcemia
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Epstein-barr virus infection and multiple sclerosis: A review. J. Neuroimmune Pharmacol. 2010, 5, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M. Smoking: Effects on multiple sclerosis susceptibility and disease progression. Ther. Adv. Neurol. Disord. 2012, 5, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Riise, T.; Casetta, I.; Drulovic, J.; Granieri, E.; Holmoy, T.; Kampman, M.T.; Landtblom, A.M.; Lauer, K.; Lossius, A.; et al. Sun exposure and multiple sclerosis risk in Norway and Italy: The EnvIMS study. Mult. Scler. 2014, 20, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L.; Simon, K.C. Vitamin D and multiple sclerosis. Lancet Neurol. 2010, 9, 599–612. [Google Scholar] [CrossRef]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef]
- Holick, M.F. Biological Effects of Sunlight, Ultraviolet Radiation, Visible Light, Infrared Radiation and Vitamin D for Health. Anticancer Res. 2016, 36, 1345–1356. [Google Scholar] [PubMed]
- Holick, M.F. Environmental factors that influence the cutaneous production of vitamin D. Am. J. Clin. Nutr. 1995, 61, 638S–645S. [Google Scholar] [CrossRef]
- Holick, M.F. Photosynthesis of vitamin D in the skin: Effect of environmental and life-style variables. Fed. Proc. 1987, 46, 1876–1882. [Google Scholar]
- Van der Mei, I.A.; Ponsonby, A.L.; Dwyer, T.; Blizzard, L.; Taylor, B.V.; Kilpatrick, T.; Butzkueven, H.; McMichael, A.J. Vitamin D levels in people with multiple sclerosis and community controls in Tasmania, Australia. J. Neurol. 2007, 254, 581–590. [Google Scholar]
- Nieves, J.; Cosman, F.; Herbert, J.; Shen, V.; Lindsay, R. High prevalence of vitamin D deficiency and reduced bone mass in multiple sclerosis. Neurology 1994, 44, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Smolders, J.; Menheere, P.; Kessels, A.; Damoiseaux, J.; Hupperts, R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult. Scler. 2008, 14, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Runia, T.F.; Hop, W.C.; de Rijke, Y.B.; Buljevac, D.; Hintzen, R.Q. Lower serum vitamin D levels are associated with a higher relapse risk in multiple sclerosis. Neurology 2012, 79, 261–266. [Google Scholar] [CrossRef]
- Mowry, E.M.; Krupp, L.B.; Milazzo, M.; Chabas, D.; Strober, J.B.; Belman, A.L.; McDonald, J.C.; Oksenberg, J.R.; Bacchetti, P.; Waubant, E. Vitamin D status is associated with relapse rate in pediatric-onset multiple sclerosis. Ann. Neurol. 2010, 67, 618–624. [Google Scholar]
- The International Multiple Sclerosis Genetics Consortium; The Wellcome Trust Case Control Consortium; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- DeLuca, H.F. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004, 80, 1689S–1696S. [Google Scholar] [CrossRef]
- Holick, M.F. The vitamin D epidemic and its health consequences. J. Nutr. 2005, 135, 2739S–2748S. [Google Scholar] [CrossRef]
- Tavera-Mendoza, L.E.; White, J.H. Cell defenses and the sunshine vitamin. Sci. Am. 2007, 297, 62–65, 68–70, 72. [Google Scholar] [CrossRef]
- Mawer, E.B.; Lumb, G.A.; Stanbury, S.W. Long biological half-life of vitamin D3 and its polar metabolites in human serum. Nature 1969, 222, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.; Goodman, D.S. The turnover and transport of vitamin D and of a polar metabolite with the properties of 25-hydroxycholecalciferol in human plasma. J. Clin. Investig. 1971, 50, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M. Vitamin D and immune function: Autocrine, paracrine or endocrine? Scand. J. Clin. Lab. Investig. Suppl. 2012, 243, 92–102. [Google Scholar]
- Overbergh, L.; Decallonne, B.; Valckx, D.; Verstuyf, A.; Depovere, J.; Laureys, J.; Rutgeerts, O.; Saint-Arnaud, R.; Bouillon, R.; Mathieu, C. Identification and immune regulation of 25-hydroxyvitamin D-1-alpha-hydroxylase in murine macrophages. Clin. Exp. Immunol. 2000, 120, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef]
- Veldman, C.M.; Cantorna, M.T.; DeLuca, H.F. Expression of 1,25-dihydroxyvitamin D(3) receptor in the immune system. Arch. Biochem. Biophys. 2000, 374, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, A.K.; Amento, E.P.; Clemens, T.L.; Holick, M.F.; Krane, S.M. Specific high-affinity receptors for 1,25-dihydroxyvitamin D3 in human peripheral blood mononuclear cells: Presence in monocytes and induction in T lymphocytes following activation. J. Clin. Endocrinol. Metab. 1983, 57, 1308–1310. [Google Scholar] [CrossRef]
- Clemens, T.L.; Garrett, K.P.; Zhou, X.Y.; Pike, J.W.; Haussler, M.R.; Dempster, D.W. Immunocytochemical localization of the 1,25-dihydroxyvitamin D3 receptor in target cells. Endocrinology 1988, 122, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.K.; Somerville, M.J.; Yoong, L.K.; Bergeron, C.; Haussler, M.R.; McLachlan, D.R. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: Correlation with calbindin-28k mRNA levels. Brain Res. Mol. Brain Res. 1992, 13, 239–250. [Google Scholar] [CrossRef]
- Neveu, I.; Naveilhan, P.; Jehan, F.; Baudet, C.; Wion, D.; De Luca, H.F.; Brachet, P. 1,25-dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Brain Res. Mol. Brain Res. 1994, 24, 70–76. [Google Scholar] [CrossRef]
- Johnson, J.A.; Grande, J.P.; Windebank, A.J.; Kumar, R. 1,25-Dihydroxyvitamin D(3) receptors in developing dorsal root ganglia of fetal rats. Brain Res. Dev. Brain Res. 1996, 92, 120–124. [Google Scholar] [CrossRef]
- Veenstra, T.D.; Prufer, K.; Koenigsberger, C.; Brimijoin, S.W.; Grande, J.P.; Kumar, R. 1,25-Dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Res. 1998, 804, 193–205. [Google Scholar] [CrossRef]
- Cornet, A.; Baudet, C.; Neveu, I.; Baron-Van Evercooren, A.; Brachet, P.; Naveilhan, P. 1,25-Dihydroxyvitamin D3 regulates the expression of VDR and NGF gene in Schwann cells in vitro. J. Neurosci. Res. 1998, 53, 742–746. [Google Scholar] [CrossRef]
- Prufer, K.; Veenstra, T.D.; Jirikowski, G.F.; Kumar, R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J. Chem. Neuroanat. 1999, 16, 135–145. [Google Scholar] [CrossRef]
- Baas, D.; Prufer, K.; Ittel, M.E.; Kuchler-Bopp, S.; Labourdette, G.; Sarlieve, L.L.; Brachet, P. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia 2000, 31, 59–68. [Google Scholar] [CrossRef]
- Langub, M.C.; Herman, J.P.; Malluche, H.H.; Koszewski, N.J. Evidence of functional vitamin D receptors in rat hippocampus. Neuroscience 2001, 104, 49–56. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D, 1st ed.; Springer Science+Business Media: New York, NY, USA, 1999; Volume XII, 458p. [Google Scholar]
- Darwish, H.; DeLuca, H.F. Vitamin D-regulated gene expression. Crit. Rev. Eukaryot. Gene Expr. 1993, 3, 89–116. [Google Scholar] [PubMed]
- Haussler, M.R.; Haussler, C.A.; Jurutka, P.W.; Thompson, P.D.; Hsieh, J.C.; Remus, L.S.; Selznick, S.H.; Whitfield, G.K. The vitamin D hormone and its nuclear receptor: Molecular actions and disease states. J. Endocrinol. 1997, 154 (Suppl. 3), S57–S73. [Google Scholar]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Hsieh, J.C.; Thompson, P.D.; Selznick, S.H.; Dominguez, C.E.; Jurutka, P.W. The nuclear vitamin D receptor: Biological and molecular regulatory properties revealed. J. Bone Miner. Res. 1998, 13, 325–349. [Google Scholar] [CrossRef]
- Rigby, W.F.; Waugh, M.; Graziano, R.F. Regulation of human monocyte HLA-DR and CD4 antigen expression, and antigen presentation by 1,25-dihydroxyvitamin D3. Blood 1990, 76, 189–197. [Google Scholar]
- Xu, H.; Soruri, A.; Gieseler, R.K.; Peters, J.H. 1,25-Dihydroxyvitamin D3 exerts opposing effects to IL-4 on MHC class-II antigen expression, accessory activity, and phagocytosis of human monocytes. Scand. J. Immunol. 1993, 38, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Almerighi, C.; Sinistro, A.; Cavazza, A.; Ciaprini, C.; Rocchi, G.; Bergamini, A. 1Alpha,25-dihydroxyvitamin D3 inhibits CD40L-induced pro-inflammatory and immunomodulatory activity in human monocytes. Cytokine 2009, 45, 190–197. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.; Cippitelli, M.; Cocciolo, M.G.; Mazzeo, D.; Di Lucia, P.; Lang, R.; Sinigaglia, F.; Panina-Bordignon, P. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J. Clin. Investig. 1998, 101, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Jonuleit, H.; Kuhn, U.; Muller, G.; Steinbrink, K.; Paragnik, L.; Schmitt, E.; Knop, J.; Enk, A.H. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 1997, 27, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Zarrabeitia, M.T.; Riancho, J.A.; Amado, J.A.; Olmos, J.M.; Gonzalez-Macias, J. Effect of calcitriol on the secretion of prostaglandin E2, interleukin 1, and tumor necrosis factor alpha by human monocytes. Bone 1992, 13, 185–189. [Google Scholar] [CrossRef]
- Kuo, Y.T.; Kuo, C.H.; Lam, K.P.; Chu, Y.T.; Wang, W.L.; Huang, C.H.; Hung, C.H. Effects of vitamin D3 on expression of tumor necrosis factor-alpha and chemokines by monocytes. J. Food Sci. 2010, 75, H200–H204. [Google Scholar] [CrossRef]
- Matilainen, J.M.; Husso, T.; Toropainen, S.; Seuter, S.; Turunen, M.P.; Gynther, P.; Yla-Herttuala, S.; Carlberg, C.; Vaisanen, S. Primary effect of 1alpha,25(OH)(2)D(3) on IL-10 expression in monocytes is short-term down-regulation. Biochim. Biophys. Acta 2010, 1803, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Gruner, S.; Brang, D.; Kampgen, E.; Lenz, A.; Trockenbacher, B.; Konwalinka, G.; Fritsch, P.O.; Steinman, R.M.; Schuler, G. Proliferating dendritic cell progenitors in human blood. J. Exp. Med. 1994, 180, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, F.; Rosenzwajg, M.; Yagello, M.; Ekman, M.; Biberfeld, P.; Gluckman, J.C. Differentiation of human dendritic cells from monocytes in vitro. Eur. J. Immunol. 1997, 27, 431–441. [Google Scholar] [CrossRef]
- Penna, G.; Adorini, L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J. Immunol. 2000, 164, 2405–2411. [Google Scholar] [CrossRef]
- Piemonti, L.; Monti, P.; Sironi, M.; Fraticelli, P.; Leone, B.E.; Dal Cin, E.; Allavena, P.; Di Carlo, V. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J. Immunol. 2000, 164, 4443–4451. [Google Scholar] [CrossRef] [PubMed]
- Van Halteren, A.G.; van Etten, E.; de Jong, E.C.; Bouillon, R.; Roep, B.O.; Mathieu, C. Redirection of human autoreactive T-cells Upon interaction with dendritic cells modulated by TX527, an analog of 1,25 dihydroxyvitamin D(3). Diabetes 2002, 51, 2119–2125. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.D.; Lutz, W.; Phan, V.A.; Bachman, L.A.; McKean, D.J.; Kumar, R. Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: A vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 6800–6805. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre d’Hellencourt, C.; Montero-Menei, C.N.; Bernard, R.; Couez, D. Vitamin D3 inhibits proinflammatory cytokines and nitric oxide production by the EOC13 microglial cell line. J. Neurosci. Res. 2003, 71, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Provvedini, D.M.; Tsoukas, C.D.; Deftos, L.J.; Manolagas, S.C. 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science 1983, 221, 1181–1183. [Google Scholar] [CrossRef]
- Provvedini, D.M.; Tsoukas, C.D.; Deftos, L.J.; Manolagas, S.C. 1 alpha,25-Dihydroxyvitamin D3-binding macromolecules in human B lymphocytes: Effects on immunoglobulin production. J. Immunol. 1986, 136, 2734–2740. [Google Scholar] [PubMed]
- Iho, S.; Takahashi, T.; Kura, F.; Sugiyama, H.; Hoshino, T. The effect of 1,25-dihydroxyvitamin D3 on in vitro immunoglobulin production in human B cells. J. Immunol. 1986, 136, 4427–4431. [Google Scholar] [PubMed]
- Lemire, J.M.; Adams, J.S.; Sakai, R.; Jordan, S.C. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J. Clin. Investig. 1984, 74, 657–661. [Google Scholar] [CrossRef]
- Nashold, F.E.; Hoag, K.A.; Goverman, J.; Hayes, C.E. Rag-1-dependent cells are necessary for 1,25-dihydroxyvitamin D(3) prevention of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2001, 119, 16–29. [Google Scholar] [CrossRef]
- Palmer, M.T.; Lee, Y.K.; Maynard, C.L.; Oliver, J.R.; Bikle, D.D.; Jetten, A.M.; Weaver, C.T. Lineage-specific effects of 1,25-dihydroxyvitamin D(3) on the development of effector CD4 T cells. J. Biol. Chem. 2011, 286, 997–1004. [Google Scholar] [CrossRef]
- Chang, S.H.; Chung, Y.; Dong, C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. J. Biol. Chem. 2010, 285, 38751–38755. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.D.; Gordon, S.A.; Cruz, J.; Cosman, F.; Cantorna, M.T. Cytokine profile in patients with multiple sclerosis following vitamin D supplementation. J. Neuroimmunol. 2003, 134, 128–132. [Google Scholar] [CrossRef]
- Jeffery, L.E.; Burke, F.; Mura, M.; Zheng, Y.; Qureshi, O.S.; Hewison, M.; Walker, L.S.; Lammas, D.A.; Raza, K.; Sansom, D.M. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J. Immunol. 2009, 183, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Thien, R.; Baier, K.; Pietschmann, P.; Peterlik, M.; Willheim, M. Interactions of 1 alpha,25-dihydroxyvitamin D3 with IL-12 and IL-4 on cytokine expression of human T lymphocytes. J. Allergy Clin. Immunol. 2005, 116, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Willheim, M.; Thien, R.; Schrattbauer, K.; Bajna, E.; Holub, M.; Gruber, R.; Baier, K.; Pietschmann, P.; Reinisch, W.; Scheiner, O.; et al. Regulatory effects of 1alpha,25-dihydroxyvitamin D3 on the cytokine production of human peripheral blood lymphocytes. J. Clin. Endocrinol. Metab. 1999, 84, 3739–3744. [Google Scholar] [PubMed]
- Bartels, L.E.; Jorgensen, S.P.; Agnholt, J.; Kelsen, J.; Hvas, C.L.; Dahlerup, J.F. 1,25-dihydroxyvitamin D3 and dexamethasone increase interleukin-10 production in CD4+ T cells from patients with Crohn’s disease. Int. Immunopharmacol. 2007, 7, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, U.; Wakita, D.; Ohkuri, T.; Chamoto, K.; Kitamura, H.; Iwakura, Y.; Nishimura, T. 1alpha,25-Dihydroxyvitamin D3 and all-trans retinoic acid synergistically inhibit the differentiation and expansion of Th17 cells. Immunol. Lett. 2010, 134, 7–16. [Google Scholar] [CrossRef]
- Colin, E.M.; Asmawidjaja, P.S.; van Hamburg, J.P.; Mus, A.M.; van Driel, M.; Hazes, J.M.; van Leeuwen, J.P.; Lubberts, E. 1,25-dihydroxyvitamin D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis. Arthritis Rheumatol. 2010, 62, 132–142. [Google Scholar] [CrossRef]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.; O’Garra, A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef]
- Khoo, A.L.; Joosten, I.; Michels, M.; Woestenenk, R.; Preijers, F.; He, X.H.; Netea, M.G.; van der Ven, A.J.; Koenen, H.J. 1,25-Dihydroxyvitamin D3 inhibits proliferation but not the suppressive function of regulatory T cells in the absence of antigen-presenting cells. Immunology 2011, 134, 459–468. [Google Scholar] [CrossRef]
- Van Belle, T.L.; Vanherwegen, A.S.; Feyaerts, D.; De Clercq, P.; Verstuyf, A.; Korf, H.; Gysemans, C.; Mathieu, C. 1,25-Dihydroxyvitamin D3 and its analog TX527 promote a stable regulatory T cell phenotype in T cells from type 1 diabetes patients. PLoS ONE 2014, 9, e109194. [Google Scholar] [CrossRef] [PubMed]
- Urry, Z.; Chambers, E.S.; Xystrakis, E.; Dimeloe, S.; Richards, D.F.; Gabrysova, L.; Christensen, J.; Gupta, A.; Saglani, S.; Bush, A.; et al. The role of 1alpha,25-dihydroxyvitamin D3 and cytokines in the promotion of distinct Foxp3+ and IL-10+ CD4+ T cells. Eur. J. Immunol. 2012, 42, 2697–2708. [Google Scholar] [CrossRef] [PubMed]
- Cippitelli, M.; Fionda, C.; Di Bona, D.; Di Rosa, F.; Lupo, A.; Piccoli, M.; Frati, L.; Santoni, A. Negative regulation of CD95 ligand gene expression by vitamin D3 in T lymphocytes. J. Immunol. 2002, 168, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Decallonne, B.; van Etten, E.; Overbergh, L.; Valckx, D.; Bouillon, R.; Mathieu, C. 1Alpha,25-dihydroxyvitamin D3 restores thymocyte apoptosis sensitivity in non-obese diabetic (NOD) mice through dendritic cells. J. Autoimmun. 2005, 24, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Pintado, C.O.; Carracedo, J.; Rodriguez, M.; Perez-Calderon, R.; Ramirez, R. 1 alpha, 25-dihydroxyvitamin D3 (calcitriol) induces apoptosis in stimulated T cells through an IL-2 dependent mechanism. Cytokine 1996, 8, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.M.; Archer, D.C. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J. Clin. Investig. 1991, 87, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Hayes, C.E.; DeLuca, H.F. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 1996, 93, 7861–7864. [Google Scholar] [CrossRef]
- Pedersen, L.B.; Nashold, F.E.; Spach, K.M.; Hayes, C.E. 1,25-dihydroxyvitamin D3 reverses experimental autoimmune encephalomyelitis by inhibiting chemokine synthesis and monocyte trafficking. J. Neurosci. Res. 2007, 85, 2480–2490. [Google Scholar] [CrossRef]
- Nashold, F.E.; Miller, D.J.; Hayes, C.E. 1,25-dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2000, 103, 171–179. [Google Scholar] [CrossRef]
- Mayne, C.G.; Spanier, J.A.; Relland, L.M.; Williams, C.B.; Hayes, C.E. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2011, 41, 822–832. [Google Scholar] [CrossRef]
- Waddell, A.; Zhao, J.; Cantorna, M.T. NKT cells can help mediate the protective effects of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis in mice. Int. Immunol. 2015, 27, 237–244. [Google Scholar] [CrossRef]
- Sotirchos, E.S.; Bhargava, P.; Eckstein, C.; Van Haren, K.; Baynes, M.; Ntranos, A.; Gocke, A.; Steinman, L.; Mowry, E.M.; Calabresi, P.A. Safety and immunologic effects of high- vs. low-dose cholecalciferol in multiple sclerosis. Neurology 2016, 86, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.S.; Liu, Y.; Gray, O.M.; Baker, J.E.; Kolbe, S.C.; Ditchfield, M.R.; Egan, G.F.; Mitchell, P.J.; Harrison, L.C.; Butzkueven, H.; et al. A randomized trial of high-dose vitamin D2 in relapsing-remitting multiple sclerosis. Neurology 2011, 77, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Loken-Amsrud, K.I.; Holmoy, T.; Bakke, S.J.; Beiske, A.G.; Bjerve, K.S.; Bjornara, B.T.; Hovdal, H.; Lilleas, F.; Midgard, R.; Pedersen, T.; et al. Vitamin D and disease activity in multiple sclerosis before and during interferon-beta treatment. Neurology 2012, 79, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Soilu-Hanninen, M.; Aivo, J.; Lindstrom, B.M.; Elovaara, I.; Sumelahti, M.L.; Farkkila, M.; Tienari, P.; Atula, S.; Sarasoja, T.; Herrala, L.; et al. A randomised, double blind, placebo controlled trial with vitamin D3 as an add on treatment to interferon beta-1b in patients with multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Stewart, N.; Simpson, S., Jr.; van der Mei, I.; Ponsonby, A.L.; Blizzard, L.; Dwyer, T.; Pittas, F.; Eyles, D.; Ko, P.; Taylor, B.V. Interferon-beta and serum 25-hydroxyvitamin D interact to modulate relapse risk in MS. Neurology 2012, 79, 254–260. [Google Scholar] [CrossRef]
- Lin, R.; Taylor, B.V.; Charlesworth, J.; van der Mei, I.; Blizzard, L.; Stewart, N.; Ponsonby, A.L.; Dwyer, T.; Pittas, F.; Simpson, S., Jr. Modulating effects of WT1 on interferon-beta-vitamin D association in MS. Acta Neurol. Scand. 2015, 131, 231–239. [Google Scholar] [CrossRef]
- Holmoy, T.; Esbensen, Q.Y.; Torkildsen, O.; Wergeland, S.; Bjerve, K.S.; Beiske, A.G.; Midgard, R.; Saltyte-Benth, J.; Hovdal, H.; Myhr, K.M. WT1 and interferon-beta-vitamin D association in MS: A longitudinal study. Acta Neurol. Scand. 2016, 133, 309–312. [Google Scholar] [CrossRef]
- Laursen, J.H.; Sondergaard, H.B.; Sorensen, P.S.; Sellebjerg, F.; Oturai, A.B. Vitamin D supplementation reduces relapse rate in relapsing-remitting multiple sclerosis patients treated with natalizumab. Mult. Scler. Relat. Disord. 2016, 10, 169–173. [Google Scholar] [CrossRef]
- Cobo-Calvo, A.; Bau, L.; Matas, E.; Romero-Pinel, L.; Mane Martinez, M.A.; Majos, C.; Martinez Yelamos, S. Effectiveness of natalizumab in patients with highly active relapsing remitting multiple sclerosis. Eur. Neurol. 2015, 73, 220–229. [Google Scholar] [CrossRef]
- Burton, J.M.; Kimball, S.; Vieth, R.; Bar-Or, A.; Dosch, H.M.; Cheung, R.; Gagne, D.; D’Souza, C.; Ursell, M.; O’Connor, P. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology 2010, 74, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://onlinelibrary.ectrims-congress.eu/ectrims/2016/32nd/147013/raymond.hupperts.high.dose.cholecalciferol.28vitamin.d329.oil.as.add-on.therapy.html (accessed on 29 December 2018).
- Muris, A.H.; Smolders, J.; Rolf, L.; Thewissen, M.; Hupperts, R.; Damoiseaux, J.; SOLARIUM Study Group. Immune regulatory effects of high dose vitamin D3 supplementation in a randomized controlled trial in relapsing remitting multiple sclerosis patients receiving IFNbeta; the SOLARIUM study. J. Neuroimmunol. 2016, 300, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Cassard, S.; Steele, S.U.; Azevedo, C.; Pelletier, D.; Sugar, E.A.; Waubant, E.; Mowry, E.M. The vitamin D to ameliorate multiple sclerosis (VIDAMS) trial: Study design for a multicenter, randomized, double-blind controlled trial of vitamin D in multiple sclerosis. Contemp. Clin. Trials 2014, 39, 288–293. [Google Scholar] [CrossRef]
- Dorr, J.; Ohlraun, S.; Skarabis, H.; Paul, F. Efficacy of vitamin D supplementation in multiple sclerosis (EVIDIMS Trial): Study protocol for a randomized controlled trial. Trials 2012, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Ponsonby, A.L.; Dear, K.; Valery, P.C.; Pender, M.P.; Taylor, B.V.; Kilpatrick, T.J.; Dwyer, T.; Coulthard, A.; Chapman, C.; et al. Sun exposure and vitamin D are independent risk factors for CNS demyelination. Neurology 2011, 76, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, N.K.; Tye, J.; Norval, M. Recent advances in urocanic acid photochemistry, photobiology and photoimmunology. Photochem. Photobiol. Sci. 2008, 7, 655–667. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. Modulation of multiple sclerosis by sunlight exposure: Role of cis-urocanic acid. J. Neuroimmunol. 2013, 261, 134–140. [Google Scholar] [CrossRef]
- Correale, J.; Ysrraelit, M.C.; Gaitan, M.I. Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain 2009, 132, 1146–1160. [Google Scholar] [CrossRef]
- Smolders, J.; Thewissen, M.; Peelen, E.; Menheere, P.; Tervaert, J.W.; Damoiseaux, J.; Hupperts, R. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS ONE 2009, 4, e6635. [Google Scholar] [CrossRef]
- Breuer, J.; Schwab, N.; Schneider-Hohendorf, T.; Marziniak, M.; Mohan, H.; Bhatia, U.; Gross, C.C.; Clausen, B.E.; Weishaupt, C.; Luger, T.A.; et al. Ultraviolet B light attenuates the systemic immune response in central nervous system autoimmunity. Ann. Neurol. 2014, 75, 739–758. [Google Scholar] [CrossRef]
- Becklund, B.R.; Severson, K.S.; Vang, S.V.; DeLuca, H.F. UV radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc. Natl. Acad. Sci. USA 2010, 107, 6418–6423. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Navid, F.; Sparwasser, T.; Clausen, B.E.; Schwarz, T. 1,25-dihydroxyvitamin D exerts similar immunosuppressive effects as UVR but is dispensable for local UVR-induced immunosuppression. J. Investig. Dermatol. 2012, 132, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Marling, S.J.; Martino, V.M.; Prahl, J.M.; Deluca, H.F. The absence of 25-hydroxyvitamin D3-1alpha-hydroxylase potentiates the suppression of EAE in mice by ultraviolet light. J. Steroid Biochem. Mol. Biol. 2016, 163, 98–102. [Google Scholar] [CrossRef]
- Tao, C.; Simpson, S., Jr.; van der Mei, I.; Blizzard, L.; Havrdova, E.; Horakova, D.; Shaygannejad, V.; Lugaresi, A.; Izquierdo, G.; Trojano, M.; et al. Higher latitude is significantly associated with an earlier age of disease onset in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Barragan, M.; Good, M.; Kolls, J.K. Regulation of Dendritic Cell Function by Vitamin D. Nutrients 2015, 7, 8127–8151. [Google Scholar] [CrossRef] [PubMed]
- Malihi, Z.; Wu, Z.; Stewart, A.W.; Lawes, C.M.; Scragg, R. Hypercalcemia, hypercalciuria, and kidney stones in long-term studies of vitamin D supplementation: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2016, 104, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.L.; Janssen, M.; de Jong, A.J. Severe hypercalcaemia syndrome with daily low-dose vitamin D supplementation. Br. J. Rheumatol. 1997, 36, 712–713. [Google Scholar] [CrossRef]
- Avenell, A.; Mak, J.C.; O’Connell, D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst. Rev. 2014, CD000227. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Wetterslev, J.; Simonetti, R.G.; Bjelakovic, M.; Gluud, C. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst. Rev. 2014, CD007470. [Google Scholar] [CrossRef]
- Vig, M.; Kinet, J.P. Calcium signaling in immune cells. Nat. Immunol. 2009, 10, 21–27. [Google Scholar] [CrossRef]
- Radermacher, A.N.; Crabtree, G.R. Monster protein controls calcium entry and fights infection. Immunity 2008, 28, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Flugel, A. Intravital real-time analysis of T-cell activation in health and disease. Cell Calcium 2017, 64, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Brand-Schieber, E.; Werner, P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp. Neurol. 2004, 189, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Parrilla, J.F.; Martinez-Moreno, M.; Gasull, X.; Mahy, N.; Rodriguez, M.J. The L-type voltage-gated calcium channel modulates microglial pro-inflammatory activity. Mol. Cell. Neurosci. 2015, 64, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Schampel, A.; Volovitch, O.; Koeniger, T.; Scholz, C.J.; Jorg, S.; Linker, R.A.; Wischmeyer, E.; Wunsch, M.; Hell, J.W.; Ergun, S.; et al. Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3295–E3304. [Google Scholar] [CrossRef] [PubMed]
- Mosayebi, G.; Haghmorad, D.; Namaki, S.; Ghazavi, A.; Ekhtiari, P.; Mirshafiey, A. Therapeutic effect of EDTA in experimental model of multiple sclerosis. Immunopharmacol. Immunotoxicol. 2010, 32, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; McCarl, C.A.; Khalil, S.; Luthy, K.; Feske, S. T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur. J. Immunol. 2010, 40, 3028–3042. [Google Scholar] [CrossRef]
- Schuhmann, M.K.; Stegner, D.; Berna-Erro, A.; Bittner, S.; Braun, A.; Kleinschnitz, C.; Stoll, G.; Wiendl, H.; Meuth, S.G.; Nieswandt, B. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J. Immunol. 2010, 184, 1536–1542. [Google Scholar] [CrossRef]
- Häusler, D.; Torke, S.; Peelen, E.; Bertsch, T.; Djukic, M.; Nau, R.; Larochelle, C.; Zamvil, S.S.; Brück, W.; Weber, M.S. Continuous high dose vitamin D exacerbates central nervous system autoimmune disease by raising T cell excitatory calcium. Brain. in revision.
- Smolders, J.; Peelen, E.; Thewissen, M.; Cohen Tervaert, J.W.; Menheere, P.; Hupperts, R.; Damoiseaux, J. Safety and T cell modulating effects of high dose vitamin D3 supplementation in multiple sclerosis. PLoS ONE 2010, 5, e15235. [Google Scholar] [CrossRef]
- Marcus, J.F.; Shalev, S.M.; Harris, C.A.; Goodin, D.S.; Josephson, S.A. Severe hypercalcemia following vitamin d supplementation in a patient with multiple sclerosis: A note of caution. Arch. Neurol. 2012, 69, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, Y.D.; Adoni, T.; Damasceno, A.; de Albuquerque Damasceno, C.A.; Ferreira, M.L.; Finkelzstejn, A.; Gomes, S.; Goncalves, M.V.; Grzesiuk, A.K.; Lins, S.; et al. Unfavorable outcomes during treatment of multiple sclerosis with high doses of vitamin D. J. Neurol. Sci. 2014, 346, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Naghavi Gargari, B.; Behmanesh, M.; Shirvani Farsani, Z.; Pahlevan Kakhki, M.; Azimi, A.R. Vitamin D supplementation up-regulates IL-6 and IL-17A gene expression in multiple sclerosis patients. Int. Immunopharmacol. 2015, 28, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Holmoy, T.; Kampman, M.T.; Smolders, J. Vitamin D in multiple sclerosis: Implications for assessment and treatment. Expert Rev. Neurother. 2012, 12, 1101–1112. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Häusler, D.; Weber, M.S. Vitamin D Supplementation in Central Nervous System Demyelinating Disease—Enough Is Enough. Int. J. Mol. Sci. 2019, 20, 218. https://doi.org/10.3390/ijms20010218
Häusler D, Weber MS. Vitamin D Supplementation in Central Nervous System Demyelinating Disease—Enough Is Enough. International Journal of Molecular Sciences. 2019; 20(1):218. https://doi.org/10.3390/ijms20010218
Chicago/Turabian StyleHäusler, Darius, and Martin S. Weber. 2019. "Vitamin D Supplementation in Central Nervous System Demyelinating Disease—Enough Is Enough" International Journal of Molecular Sciences 20, no. 1: 218. https://doi.org/10.3390/ijms20010218
APA StyleHäusler, D., & Weber, M. S. (2019). Vitamin D Supplementation in Central Nervous System Demyelinating Disease—Enough Is Enough. International Journal of Molecular Sciences, 20(1), 218. https://doi.org/10.3390/ijms20010218