The Interaction between Viral and Environmental Risk Factors in the Pathogenesis of Multiple Sclerosis

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
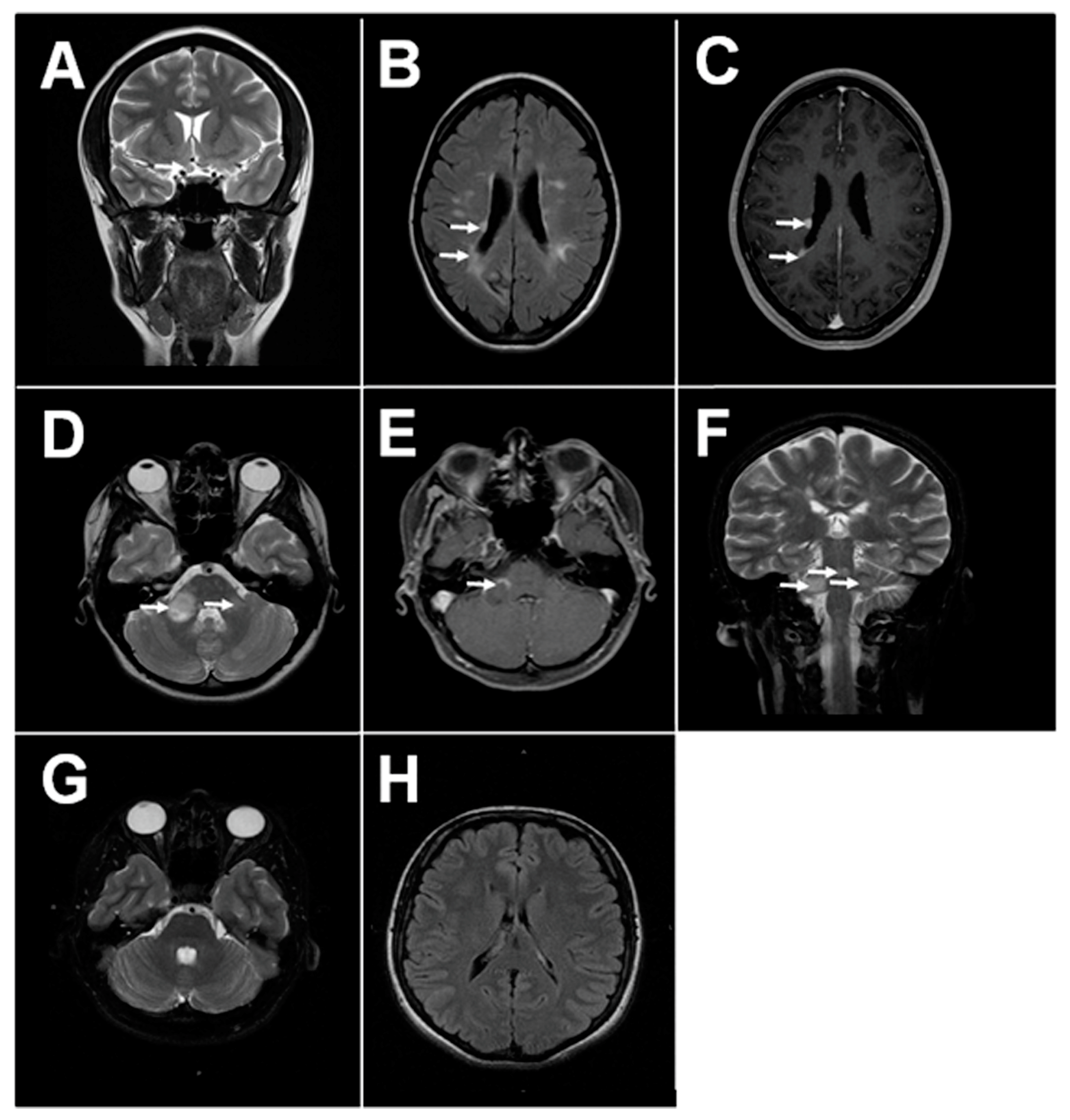
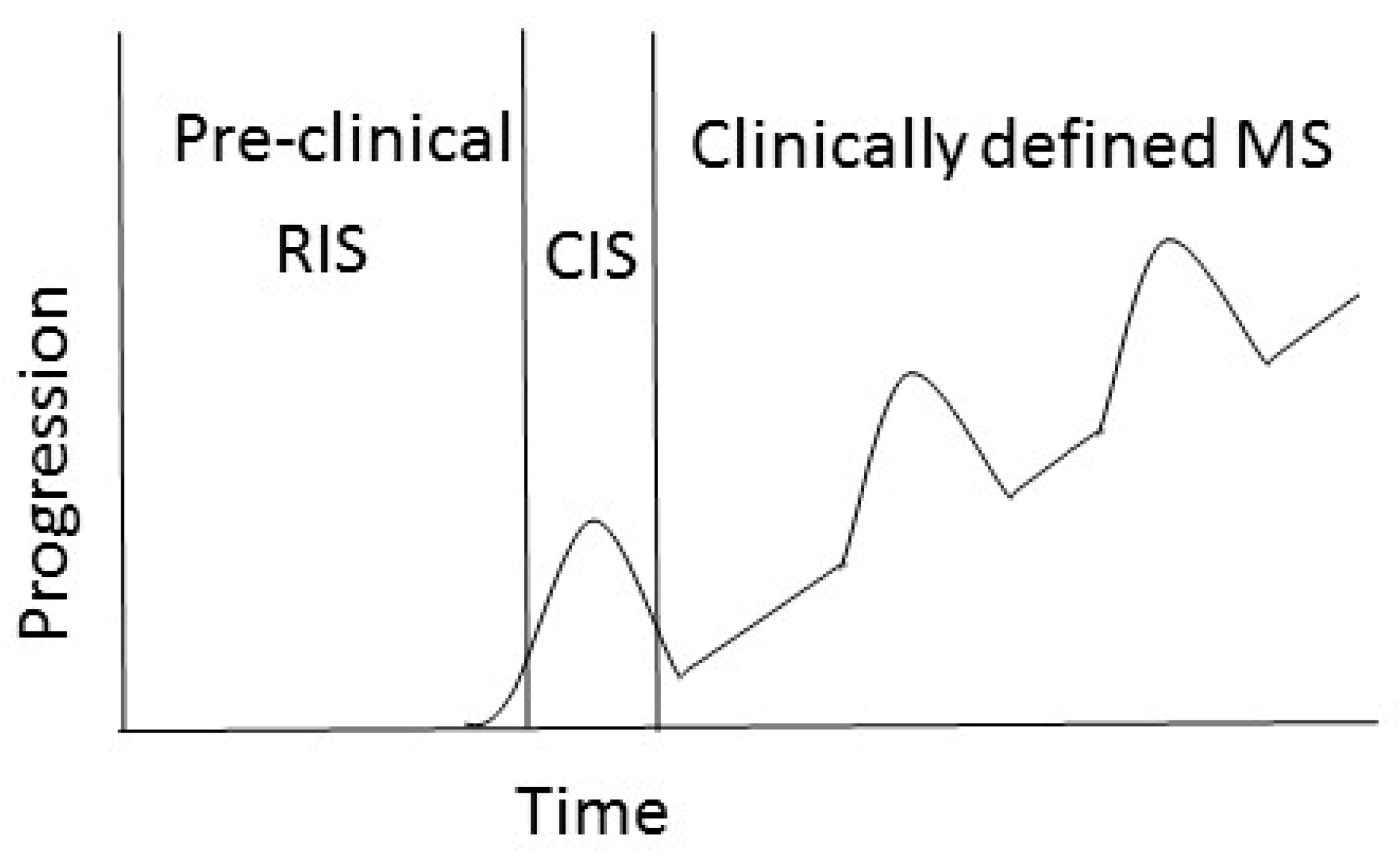
1.1. MS Pathology and Clinical Disease
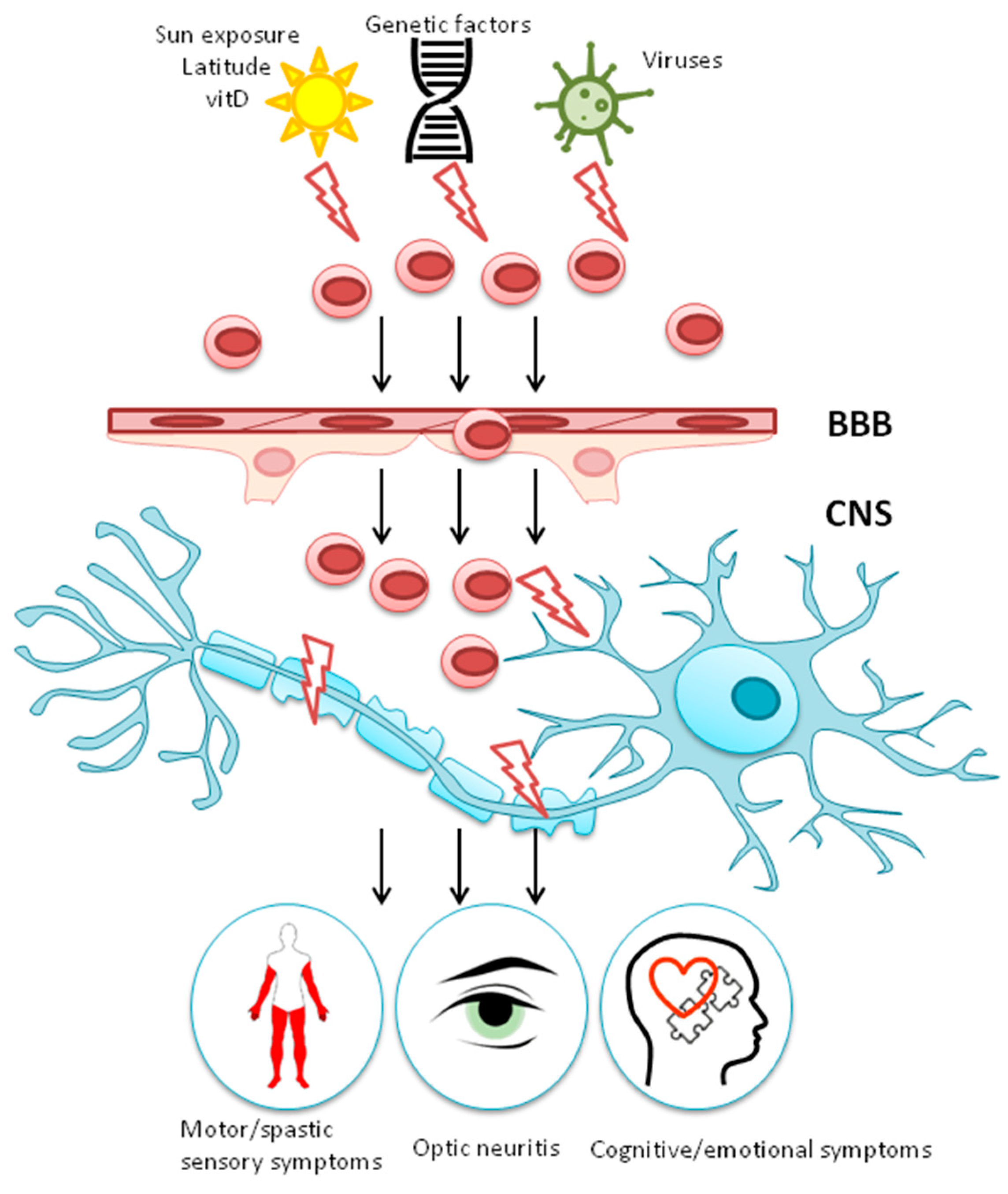
1.2. Gene-Environment Interaction
2. Environmental Risk Factors for MS
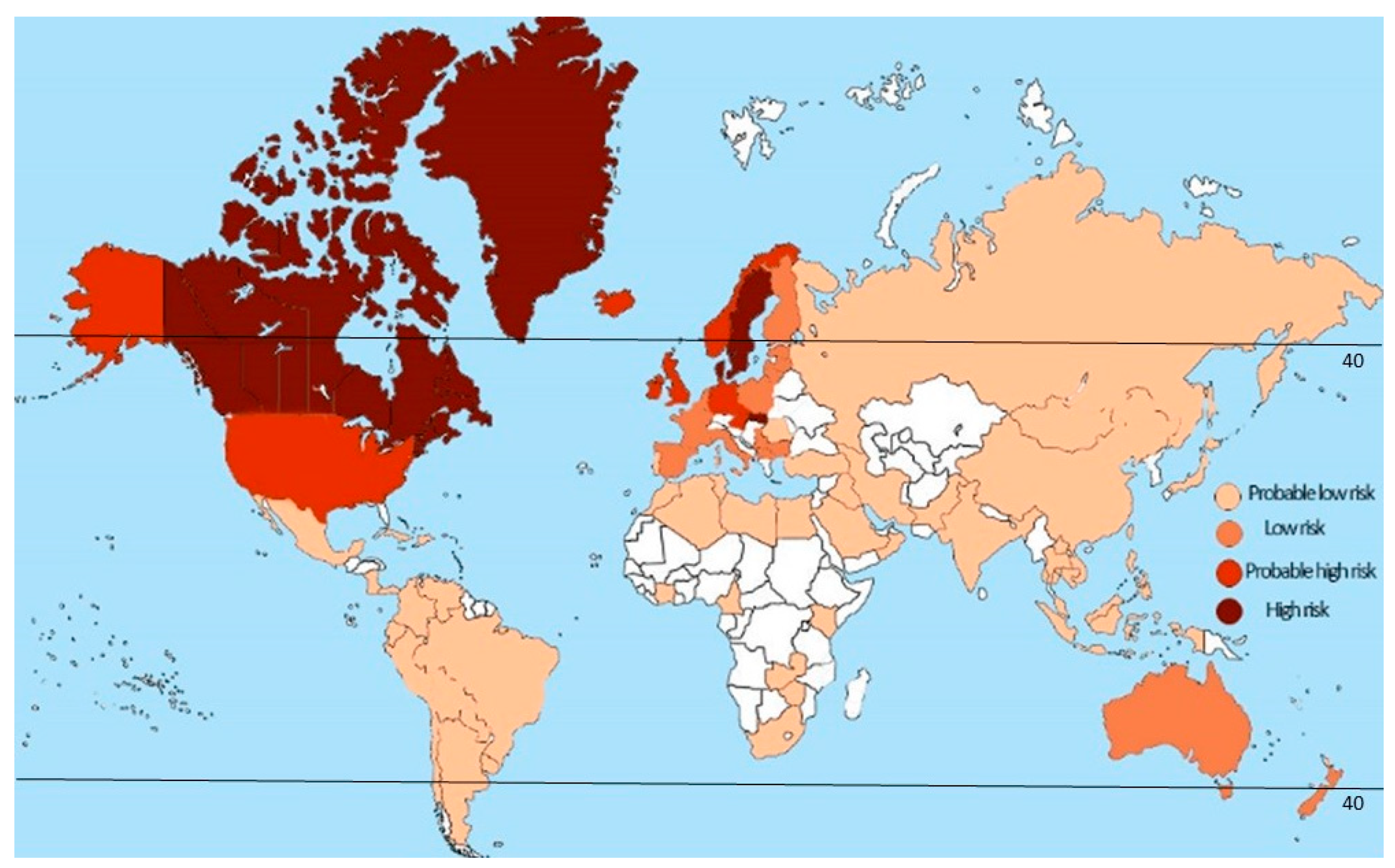
2.1. Latitude
2.2. vitD
2.3. Lifestyle Factors
2.4. Viruses and Other Infecitous Agents
2.5. Endogenous Retrovirus Expression
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Continuum (Minneap Minn) 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.L.; Kaplan, R.D.; Grewe, G.; White, R.T.; Salberg, L.M. The ovoid lesion: A new MR observation in patients with multiple sclerosis. AJNR Am. J. Neuroradiol. 1989, 10, 303–305. [Google Scholar] [PubMed]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Calabrese, M.; Favaretto, A.; Martini, V.; Gallo, P. Grey matter lesions in MS: From histology to clinical implications. Prion 2013, 7, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; De Stefano, N.; Atzori, M.; Bernardi, V.; Mattisi, I.; Barachino, L.; Morra, A.; Rinaldi, L.; Romualdi, C.; Perini, P.; et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch. Neurol. 2007, 64, 1416–1422. [Google Scholar] [CrossRef]
- Grossman, R.I.; McGowan, J.C. Perspectives on multiple sclerosis. AJNR Am. J. Neuroradiol. 1998, 19, 1251–1265. [Google Scholar]
- Ge, Y. Multiple sclerosis: The role of MR imaging. AJNR Am. J. Neuroradiol. 2006, 27, 1165–1176. [Google Scholar]
- Planas, R.; Metz, I.; Ortiz, Y.; Vilarrasa, N.; Jelcic, I.; Salinas-Riester, G.; Heesen, C.; Bruck, W.; Martin, R.; Sospedra, M. Central role of Th2/Tc2 lymphocytes in pattern II multiple sclerosis lesions. Ann. Clin. Transl. Neurol. 2015, 2, 875–893. [Google Scholar] [CrossRef]
- Fritzsching, B.; Haas, J.; Konig, F.; Kunz, P.; Fritzsching, E.; Poschl, J.; Krammer, P.H.; Bruck, W.; Suri-Payer, E.; Wildemann, B. Intracerebral human regulatory T cells: Analysis of CD4+ CD25+ FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. PLoS ONE 2011, 6, e17988. [Google Scholar] [CrossRef]
- Woodroofe, M.N.; Bellamy, A.S.; Feldmann, M.; Davison, A.N.; Cuzner, M.L. Immunocytochemical characterisation of the immune reaction in the central nervous system in multiple sclerosis. Possible role for microglia in lesion growth. J. Neurol. Sci. 1986, 74, 135–152. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Capello, E.; Mancardi, G.L.; Aloisi, F. Dendritic cells in multiple sclerosis lesions: Maturation stage, myelin uptake, and interaction with proliferating T cells. J. Neuropathol. Exp. Neurol. 2006, 65, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Al-Asmi, A.; Duquette, P.; Antel, J.P. Lymphocyte migration and multiple sclerosis: Relation with disease course and therapy. Ann. Neurol. 1999, 46, 253–256. [Google Scholar] [CrossRef]
- Washington, R.; Burton, J.; Todd, R.F., 3rd; Newman, W.; Dragovic, L.; Dore-Duffy, P. Expression of immunologically relevant endothelial cell activation antigens on isolated central nervous system microvessels from patients with multiple sclerosis. Ann. Neurol. 1994, 35, 89–97. [Google Scholar] [CrossRef]
- Man, S.; Tucky, B.; Bagheri, N.; Li, X.; Kochar, R.; Ransohoff, R.M. alpha4 Integrin/FN-CS1 mediated leukocyte adhesion to brain microvascular endothelial cells under flow conditions. J. Neuroimmunol. 2009, 210, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J. Neural Transm. (Vienna) 2006, 113, 477–485. [Google Scholar] [CrossRef]
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995, 37, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Pelfrey, C.M.; Tranquill, L.R.; Maloni, H.; McFarland, H.F. VLA-4 expression on peripheral blood lymphocytes is downregulated after treatment of multiple sclerosis with interferon beta. Neurology 1997, 49, 1111–1116. [Google Scholar] [CrossRef]
- Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O’Connor, P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003, 348, 15–23. [Google Scholar] [CrossRef]
- Stuve, O.; Marra, C.M.; Jerome, K.R.; Cook, L.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Phillips, J.T.; Arendt, G.; Hemmer, B.; et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. 2006, 59, 743–747. [Google Scholar] [CrossRef]
- De Andres, C.; Teijeiro, R.; Alonso, B.; Sanchez-Madrid, F.; Martinez, M.L.; Guzman de Villoria, J.; Fernandez-Cruz, E.; Sanchez-Ramon, S. Long-term decrease in VLA-4 expression and functional impairment of dendritic cells during natalizumab therapy in patients with multiple sclerosis. PLoS ONE 2012, 7, e34103. [Google Scholar] [CrossRef]
- Stuve, O.; Marra, C.M.; Bar-Or, A.; Niino, M.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Phillips, J.T.; Arendt, G.; Jerome, K.R.; et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch. Neurol. 2006, 63, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Uhm, J.H.; Dooley, N.P.; Stuve, O.; Francis, G.S.; Duquette, P.; Antel, J.P.; Yong, V.W. Migratory behavior of lymphocytes isolated from multiple sclerosis patients: Effects of interferon beta-1b therapy. Ann. Neurol. 1999, 46, 319–324. [Google Scholar] [CrossRef]
- Stuve, O.; Dooley, N.P.; Uhm, J.H.; Antel, J.P.; Francis, G.S.; Williams, G.; Yong, V.W. Interferon beta-1b decreases the migration of T lymphocytes in vitro: Effects on matrix metalloproteinase-9. Ann. Neurol. 1996, 40, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Biernacki, K.; Lavoie, J.F.; Poirier, J.; Duquette, P.; Antel, J.P. Migration of multiple sclerosis lymphocytes through brain endothelium. Arch. Neurol. 2002, 59, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef]
- Olsson, T.; Zhi, W.W.; Hojeberg, B.; Kostulas, V.; Jiang, Y.P.; Anderson, G.; Ekre, H.P.; Link, H. Autoreactive T lymphocytes in multiple sclerosis determined by antigen-induced secretion of interferon-gamma. J. Clin. Investig. 1990, 86, 981–985. [Google Scholar] [CrossRef]
- Wheeler, R.D.; Zehntner, S.P.; Kelly, L.M.; Bourbonniere, L.; Owens, T. Elevated interferon gamma expression in the central nervous system of tumour necrosis factor receptor 1-deficient mice with experimental autoimmune encephalomyelitis. Immunology 2006, 118, 527–538. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Dos Passos, G.R.; Sato, D.K.; Becker, J.; Fujihara, K. Th17 Cells Pathways in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders: Pathophysiological and Therapeutic Implications. Mediat. Inflamm. 2016, 2016, 5314541. [Google Scholar] [CrossRef] [PubMed]
- Kuenz, B.; Lutterotti, A.; Ehling, R.; Gneiss, C.; Haemmerle, M.; Rainer, C.; Deisenhammer, F.; Schocke, M.; Berger, T.; Reindl, M. Cerebrospinal fluid B cells correlate with early brain inflammation in multiple sclerosis. PLoS ONE 2008, 3, e2559. [Google Scholar] [CrossRef]
- Freedman, M.S.; Thompson, E.J.; Deisenhammer, F.; Giovannoni, G.; Grimsley, G.; Keir, G.; Ohman, S.; Racke, M.K.; Sharief, M.; Sindic, C.J.; et al. Recommended standard of cerebrospinal fluid analysis in the diagnosis of multiple sclerosis: A consensus statement. Arch. Neurol. 2005, 62, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.; Svenningsson, R.; Alping, P.; Novakova, L.; Bjorck, A.; Fink, K.; Islam-Jakobsson, P.; Malmestrom, C.; Axelsson, M.; Vagberg, M.; et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology 2016, 87, 2074–2081. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.; De Seze, J.; Montalban, X.; Selmaj, K.; et al. Ofatumumab versus teriflunomide in relapsing MS: Adaptive design of two Phase 3 studies (ASCLEPIOS I and ASCLEPIOS II). Neurology 2017, 18, 88. [Google Scholar]
- Genmab Announces Phase III Studies of Ofatumumab in Relapsing Multiple Sclerosis. Available online: http://ir.genmab.com/releasedetail.cfm?releaseid=973889 (accessed on 2 June 2016).
- Castillo-Trivino, T.; Braithwaite, D.; Bacchetti, P.; Waubant, E. Rituximab in relapsing and progressive forms of multiple sclerosis: A systematic review. PLoS ONE 2013, 8, e66308. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.P.; Barnett, M.H.; Parratt, J.D.; Prineas, J.W. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 2009, 66, 739–753. [Google Scholar] [CrossRef]
- Schonrock, L.M.; Kuhlmann, T.; Adler, S.; Bitsch, A.; Bruck, W. Identification of glial cell proliferation in early multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 1998, 24, 320–330. [Google Scholar] [CrossRef]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Markovic-Plese, S.; Lacet, B.; Raus, J.; Weiner, H.L.; Hafler, D.A. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J. Exp. Med. 1994, 179, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Parker, K.C.; Turner, R.V.; McFarland, H.F.; Coligan, J.E.; Biddison, W.E. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc. Natl. Acad. Sci. USA 1994, 91, 10859–10863. [Google Scholar] [CrossRef] [PubMed]
- Huseby, E.S.; Liggitt, D.; Brabb, T.; Schnabel, B.; Ohlen, C.; Goverman, J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 2001, 194, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Biegler, B.W.; Yan, S.X.; Ortega, S.B.; Tennakoon, D.K.; Racke, M.K.; Karandikar, N.J. Clonal composition of neuroantigen-specific CD8+ and CD4+ T-cells in multiple sclerosis. J. Neuroimmunol. 2011, 234, 131–140. [Google Scholar] [CrossRef] [PubMed]
- De Paula Alves Sousa, A.; Johnson, K.R.; Nicholas, R.; Darko, S.; Price, D.A.; Douek, D.C.; Jacobson, S.; Muraro, P.A. Intrathecal T-cell clonal expansions in patients with multiple sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Vassall, K.A.; Bamm, V.V.; Harauz, G. MyelStones: The executive roles of myelin basic protein in myelin assembly and destabilization in multiple sclerosis. Biochem. J. 2015, 472, 17–32. [Google Scholar] [CrossRef]
- Noseworthy, J.H. Progress in determining the causes and treatment of multiple sclerosis. Nature 1999, 399, A40–A47. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Koch, M.; Kingwell, E.; Rieckmann, P.; Tremlett, H. The natural history of primary progressive multiple sclerosis. Neurology 2009, 73, 1996–2002. [Google Scholar] [CrossRef]
- Khaleeli, Z.; Ciccarelli, O.; Manfredonia, F.; Barkhof, F.; Brochet, B.; Cercignani, M.; Dousset, V.; Filippi, M.; Montalban, X.; Polman, C.; et al. Predicting progression in primary progressive multiple sclerosis: A 10-year multicenter study. Ann. Neurol. 2008, 63, 790–793. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, M.M.; Geurts, J.J.; Rostrup, E.; Uitdehaag, B.M.; Polman, C.H.; Barkhof, F.; Vrenken, H. Clinical correlations of brain lesion distribution in multiple sclerosis. J. Magn. Reson. Imaging 2009, 29, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Wegner, C.; Bruck, W. Inflammation, demyelination, and degeneration—Recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282. [Google Scholar] [CrossRef]
- Richards, R.G.; Sampson, F.C.; Beard, S.M.; Tappenden, P. A review of the natural history and epidemiology of multiple sclerosis: Implications for resource allocation and health economic models. Health Technol Assess 2002, 6, 1–73. [Google Scholar] [CrossRef]
- Sumowski, J.F.; Benedict, R.; Enzinger, C.; Filippi, M.; Geurts, J.J.; Hamalainen, P.; Hulst, H.; Inglese, M.; Leavitt, V.M.; Rocca, M.A.; et al. Cognition in multiple sclerosis: State of the field and priorities for the future. Neurology 2018, 90, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Guidelines for preventing and treating vitamin D deficiency and insufficiency revisited. J. Clin. Endocrinol. Metab. 2012, 97, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Binzer, M.; Forsgren, L.; Holmgren, G.; Drugge, U.; Fredrikson, S. Familial clustering of multiple sclerosis in a northern Swedish rural district. J. Neurol. Neurosurg. Psychiatry 1994, 57, 497–499. [Google Scholar] [CrossRef]
- Tienari, P.J.; Sumelahti, M.L.; Rantamaki, T.; Wikstrom, J. Multiple sclerosis in western Finland: Evidence for a founder effect. Clin. Neurol. Neurosurg. 2004, 106, 175–179. [Google Scholar] [CrossRef]
- Nicoletti, A.; Lo Fermo, S.; Reggio, E.; Tarantello, R.; Liberto, A.; Le Pira, F.; Patti, F.; Reggio, A. A possible spatial and temporal cluster of multiple sclerosis in the town of Linguaglossa, Sicily. J. Neurol. 2005, 252, 921–925. [Google Scholar] [CrossRef]
- Sharpe, G.; Price, S.E.; Last, A.; Thompson, R.J. Multiple sclerosis in island populations: Prevalence in the Bailiwicks of Guernsey and Jersey. J. Neurol. Neurosurg. Psychiatry 1995, 58, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Charlton, D. High incidence and prevalence of multiple sclerosis in south east Scotland: Evidence of a genetic predisposition. J. Neurol. Neurosurg. Psychiatry 1998, 64, 730–735. [Google Scholar] [CrossRef]
- Gray, O.M.; McDonnell, G.V.; Hawkins, S.A. Factors in the rising prevalence of multiple sclerosis in the north-east of Ireland. Mult. Scler. 2008, 14, 880–886. [Google Scholar] [CrossRef]
- Smestad, C.; Sandvik, L.; Holmoy, T.; Harbo, H.F.; Celius, E.G. Marked differences in prevalence of multiple sclerosis between ethnic groups in Oslo, Norway. J. Neurol. 2008, 255, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Harbo, H.F.; Utsi, E.; Lorentzen, A.R.; Kampman, M.T.; Celius, E.G.; Myhr, K.M.; Lie, B.A.; Mellgren, S.I.; Thorsby, E. Low frequency of the disease-associated DRB1*15-DQB1*06 haplotype may contribute to the low prevalence of multiple sclerosis in Sami. Tissue Antigens 2007, 69, 299–304. [Google Scholar] [CrossRef]
- Dean, G.; Elian, M.; de Bono, A.G.; Asciak, R.P.; Vella, N.; Mifsud, V.; Aquilina, J. Multiple sclerosis in Malta in 1999: An update. J. Neurol. Neurosurg. Psychiatry 2002, 73, 256–260. [Google Scholar] [CrossRef]
- Willer, C.J.; Dyment, D.A.; Risch, N.J.; Sadovnick, A.D.; Ebers, G.C. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12877–12882. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.G.; Rose, J.W.; Smoker, W.; Petajan, J.H. MRI in familial multiple sclerosis. Neurology 1990, 40, 900–903. [Google Scholar] [CrossRef]
- Tienari, P.J.; Salonen, O.; Wikstrom, J.; Valanne, L.; Palo, J. Familial multiple sclerosis: MRI findings in clinically affected and unaffected siblings. J. Neurol. Neurosurg. Psychiatry 1992, 55, 883–886. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Didonna, A.; Oksenberg, J.R. The Genetics of Multiple Sclerosis; Codon Publications: Philadelphia, PA, USA, 2017. [Google Scholar]
- Krause-Kyora, B.; Nutsua, M.; Boehme, L.; Pierini, F.; Pedersen, D.D.; Kornell, S.C.; Drichel, D.; Bonazzi, M.; Mobus, L.; Tarp, P.; et al. Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans. Nat. Commun. 2018, 9, 1569. [Google Scholar] [CrossRef]
- Petrone, A.; Battelino, T.; Krzisnik, C.; Bugawan, T.; Erlich, H.; Di Mario, U.; Pozzilli, P.; Buzzetti, R. Similar incidence of type 1 diabetes in two ethnically different populations (Italy and Slovenia) is sustained by similar HLA susceptible/protective haplotype frequencies. Tissue Antigens 2002, 60, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Kular, L.; Liu, Y.; Ruhrmann, S.; Zheleznyakova, G.; Marabita, F.; Gomez-Cabrero, D.; James, T.; Ewing, E.; Linden, M.; Gornikiewicz, B.; et al. DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis. Nat. Commun. 2018, 9, 2397. [Google Scholar] [CrossRef] [PubMed]
- Briggs, F.B.S.; Yu, J.C.; Davis, M.F.; Jiangyang, J.; Fu, S.; Parrotta, E.; Gunzler, D.D.; Ontaneda, D. Multiple sclerosis risk factors contribute to onset heterogeneity. Mult. Scler. Relat. Disord. 2018, 28, 11–16. [Google Scholar] [CrossRef]
- Maver, A.; Lavtar, P.; Ristic, S.; Stopinsek, S.; Simcic, S.; Hocevar, K.; Sepcic, J.; Drulovic, J.; Pekmezovic, T.; Novakovic, I.; et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef] [PubMed]
- Traboulsee, A.L.; Sadovnick, A.D.; Encarnacion, M.; Bernales, C.Q.; Yee, I.M.; Criscuoli, M.G.; Vilarino-Guell, C. Common genetic etiology between “multiple sclerosis-like” single-gene disorders and familial multiple sclerosis. Hum. Genet. 2017, 136, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Colomba, P.; Zizzo, C.; Alessandro, R.; Cammarata, G.; Scalia, S.; Giordano, A.; Pieroni, M.; Sicurella, L.; Amico, L.; Burlina, A.; et al. Fabry disease and multiple sclerosis misdiagnosis: The role of family history and neurological signs. Oncotarget 2018, 9, 7758–7762. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Handel, A.E.; Giovannoni, G.; Rutherford Siegel, S.; Ebers, G.C.; Chaplin, G. Relationship of UV exposure to prevalence of multiple sclerosis in England. Neurology 2011, 76, 1410–1414. [Google Scholar] [CrossRef]
- Donnan, P.T.; Parratt, J.D.; Wilson, S.V.; Forbes, R.B.; O’Riordan, J.I.; Swingler, R.J. Multiple sclerosis in Tayside, Scotland: Detection of clusters using a spatial scan statistic. Mult. Scler. (Houndmills, Basingstoke, England) 2005, 11, 403–408. [Google Scholar] [CrossRef]
- Simpson, S., Jr.; Blizzard, L.; Otahal, P.; Van der Mei, I.; Taylor, B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1132–1141. [Google Scholar] [CrossRef]
- Fromont, A.; Binquet, C.; Sauleau, E.A.; Fournel, I.; Bellisario, A.; Adnet, J.; Weill, A.; Vukusic, S.; Confavreux, C.; Debouverie, M.; et al. Geographic variations of multiple sclerosis in France. Brain 2010, 133, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Benjaminsen, E.; Olavsen, J.; Karlberg, M.; Alstadhaug, K.B. Multiple sclerosis in the far north--incidence and prevalence in Nordland County, Norway, 1970-2010. BMC Neurol. 2014, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Risco, J.; Maldonado, H.; Luna, L.; Osada, J.; Ruiz, P.; Juarez, A.; Vizcarra, D. Latitudinal prevalence gradient of multiple sclerosis in Latin America. Mult. Scler. (Houndmills, Basingstoke, England) 2011, 17, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Swingler, R.J.; Compston, D. The distribution of multiple sclerosis in the United Kingdom. J. Neurol Neurosurg. Psychiatry 1986, 49, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.M. Observations on the prevalence of multiple sclerosis in Northern Scotland. Brain 1956, 79, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Chandraratna, D.; Angood, C.; Tremlett, H.; Baker, C.; Taylor, B.V.; Thompson, A.J. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology 2014, 83, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, P. Multiple sclerosis: Vitamin D and calcium as environmental determinants of prevalence. Int. J. Environ. Stud. 1973, 6, 19–27. [Google Scholar] [CrossRef]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Stamnes, K.; Volden, G.; Falk, E.S. Ultraviolet radiation at high latitudes and the risk of skin cancer. Photodermatology 1989, 6, 110–117. [Google Scholar]
- Langston, M.; Dennis, L.; Lynch, C.; Roe, D.; Brown, H. Temporal Trends in Satellite-Derived Erythemal UVB and Implications for Ambient Sun Exposure Assessment. Int. J. Environ. Res. Public Health. 2017, 14, 176. [Google Scholar] [CrossRef]
- Spina, C.; Tangpricha, V.; Yao, M.; Zhou, W.; Wolfe, M.M.; Maehr, H.; Uskokovic, M.; Adorini, L.; Holick, M.F. Colon cancer and solar ultraviolet B radiation and prevention and treatment of colon cancer in mice with vitamin D and its Gemini analogs. J. Steroid Biochem. Mol. Biol. 2005, 97, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Ladizesky, M.; Lu, Z.; Oliveri, B.; San Roman, N.; Diaz, S.; Holick, M.F.; Mautalen, C. Solar ultraviolet B radiation and photoproduction of vitamin D3 in central and southern areas of Argentina. J. Bone Miner Res. 1995, 10, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.R.; Engelsen, O. Calculated ultraviolet exposure levels for a healthy vitamin D status. Photochem. Photobiol. 2006, 82, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Norval, M.; Halliday, G.M. The consequences of UV-induced immunosuppression for human health. Photochem. Photobiol. 2011, 87, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Marling, S.J.; Beaver, E.F.; Severson, K.S.; Deluca, H.F. UV light selectively inhibits spinal cord inflammation and demyelination in experimental autoimmune encephalomyelitis. Arch. Biochem. Biophys. 2015, 567, 75–82. [Google Scholar] [CrossRef]
- Becklund, B.R.; Severson, K.S.; Vang, S.V.; DeLuca, H.F. UV radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc. Natl. Acad. Sci. USA 2010, 107, 6418–6423. [Google Scholar] [CrossRef]
- Nieves, J.; Cosman, F.; Herbert, J.; Shen, V.; Lindsay, R. High prevalence of vitamin D deficiency and reduced bone mass in multiple sclerosis. Neurology 1994, 44, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Brola, W.; Sobolewski, P.; Szczuchniak, W.; Goral, A.; Fudala, M.; Przybylski, W.; Opara, J. Association of seasonal serum 25-hydroxyvitamin D levels with disability and relapses in relapsing-remitting multiple sclerosis. Eur. J. Clin. Nutr. 2016, 70, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Kepczynska, K.; Zajda, M.; Lewandowski, Z.; Przedlacki, J.; Zakrzewska-Pniewska, B. Bone metabolism and vitamin D status in patients with multiple sclerosis. Neurol. Neurochir. Pol. 2016, 50, 251–257. [Google Scholar]
- Pandit, L.; Ramagopalan, S.V.; Malli, C.; D’Cunha, A.; Kunder, R.; Shetty, R. Association of vitamin D and multiple sclerosis in India. Mult. Scler. (Houndmills, Basingstoke, England) 2013, 19, 1592–1596. [Google Scholar] [CrossRef]
- Gianfrancesco, M.A.; Stridh, P.; Rhead, B.; Shao, X.; Xu, E.; Graves, J.S.; Chitnis, T.; Waldman, A.; Lotze, T.; Schreiner, T.; et al. Evidence for a causal relationship between low vitamin D, high BMI, and pediatric-onset MS. Neurology 2017, 88, 1623–1629. [Google Scholar] [CrossRef]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef]
- Salzer, J.; Hallmans, G.; Nystrom, M.; Stenlund, H.; Wadell, G.; Sundstrom, P. Vitamin D as a protective factor in multiple sclerosis. Neurology 2012, 79, 2140–2145. [Google Scholar] [CrossRef]
- Ross, A.C.; Manson, J.E.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J. Clin. Endocrinol. Metab. 2011, 96, 53–58. [Google Scholar] [CrossRef]
- Chapuy, M.C.; Preziosi, P.; Maamer, M.; Arnaud, S.; Galan, P.; Hercberg, S.; Meunier, P.J. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos. Int. 1997, 7, 439–443. [Google Scholar] [CrossRef]
- Kasahara, A.K.; Singh, R.J.; Noymer, A. Vitamin D (25OHD) Serum Seasonality in the United States. PLoS ONE 2013, 8, e65785. [Google Scholar] [CrossRef]
- Heidari, B.; Haji Mirghassemi, M.B. Seasonal variations in serum vitamin D according to age and sex. Caspian J. Intern. Med. 2012, 3, 535–540. [Google Scholar] [PubMed]
- Dobson, R.; Giovannoni, G.; Ramagopalan, S. The month of birth effect in multiple sclerosis: Systematic review, meta-analysis and effect of latitude. J. Neurol. Neurosurg. Psychiatry 2013, 84, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Dyment, D.A.; Sadovnick, A.D.; Rothwell, P.M.; Murray, T.J.; Ebers, G.C. Timing of birth and risk of multiple sclerosis: Population based study. BMJ 2005, 330, 120. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.H.; Sondergaard, H.B.; Sorensen, P.S.; Sellebjerg, F.; Oturai, A.B. Vitamin D supplementation reduces relapse rate in relapsing-remitting multiple sclerosis patients treated with natalizumab. Mult. Scler. Relat. Disord. 2016, 10, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ashtari, F.; Toghianifar, N.; Zarkesh-Esfahani, S.H.; Mansourian, M. High dose Vitamin D intake and quality of life in relapsing-remitting multiple sclerosis: A randomized, double-blind, placebo-controlled clinical trial. Neurol. Res. 2016, 38, 888–892. [Google Scholar] [CrossRef]
- Myhr, K.M. Vitamin D treatment in multiple sclerosis. J. Neurol. Sci 2009, 286, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Jansson, B.; Bonelli, F. Modulation of inflammatory and immune responses by vitamin D. J. Autoimmun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Harding, K.; Tilling, K.; MacIver, C.; Willis, M.; Joseph, F.; Ingram, G.; Hirst, C.; Wardle, M.; Pickersgill, T.; Ben-Shlomo, Y.; et al. Seasonal variation in multiple sclerosis relapse. J. Neurol. 2017, 264, 1059–1067. [Google Scholar] [CrossRef]
- Katz Sand, I. The Role of Diet in Multiple Sclerosis: Mechanistic Connections and Current Evidence. Curr. Nutr. Rep. 2018, 7, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Degelman, M.L.; Herman, K.M. Smoking and multiple sclerosis: A systematic review and meta-analysis using the Bradford Hill criteria for causation. Mult. Scler. Relat. Disord. 2017, 17, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Rasul, T.; Frederiksen, J.L. Link between overweight/obese in children and youngsters and occurrence of multiple sclerosis. J. Neurol. 2018, 265, 2755–2763. [Google Scholar] [CrossRef]
- Melbye, P.; Olsson, A.; Hansen, T.H.; Sondergaard, H.B.; Bang Oturai, A. Short-chain fatty acids and gut microbiota in multiple sclerosis. Acta Neurol. Scand. 2018. [Google Scholar] [CrossRef]
- Barragan-Martinez, C.; Speck-Hernandez, C.A.; Montoya-Ortiz, G.; Mantilla, R.D.; Anaya, J.M.; Rojas-Villarraga, A. Organic solvents as risk factor for autoimmune diseases: A systematic review and meta-analysis. PLoS ONE 2012, 7, e51506. [Google Scholar] [CrossRef]
- Kotzamani, D.; Panou, T.; Mastorodemos, V.; Tzagournissakis, M.; Nikolakaki, H.; Spanaki, C.; Plaitakis, A. Rising incidence of multiple sclerosis in females associated with urbanization. Neurology 2012, 78, 1728–1735. [Google Scholar] [CrossRef]
- Hussain, M.S.; Tripathi, V. Smoking under hypoxic conditions: A potent environmental risk factor for inflammatory and autoimmune diseases. Mil. Med. Res. 2018, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gomez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Epstein-barr virus infection and multiple sclerosis: A review. J. Neuroimmune Pharmacol. 2010, 5, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between Epstein-Barr virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist 2011, 17, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. 2007, 204, 2899–2912. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Wu, J.; Lucas, R.; Smith, J.; Gonzales, E.; Amezcua, L.; Haraszti, S.; Chen, L.H.; Quach, H.; James, J.A.; et al. Epstein-Barr virus, cytomegalovirus, and multiple sclerosis susceptibility: A multiethnic study. Neurology 2017, 89, 1330–1337. [Google Scholar] [CrossRef]
- Makhani, N.; Banwell, B.; Tellier, R.; Yea, C.; McGovern, S.; O’Mahony, J.; Ahorro, J.M.; Arnold, D.; Sadovnick, A.D.; Marrie, R.A.; et al. Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult. Scler. 2016, 22, 385–388. [Google Scholar] [CrossRef]
- Clerico, M.; De Mercanti, S.; Artusi, C.A.; Durelli, L.; Naismith, R.T. Active CMV infection in two patients with multiple sclerosis treated with alemtuzumab. Mult. Scler. 2017, 23, 874–876. [Google Scholar] [CrossRef]
- Vanheusden, M.; Stinissen, P.; t Hart, B.A.; Hellings, N. Cytomegalovirus: A culprit or protector in multiple sclerosis? Trends Mol. Med. 2015, 21, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kira, J.I.; Isobe, N. Helicobacter pylori infection and demyelinating disease of the central nervous system. J. Neuroimmunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Koskderelioglu, A.; Afsar, I.; Pektas, B.; Gedizlioglu, M. Is Toxoplasma gondii infection protective against multiple sclerosis risk? Mult. Scler. Relat. Disord. 2017, 15, 7–10. [Google Scholar] [CrossRef]
- Dixit, A.; Tanaka, A.; Greer, J.M.; Donnelly, S. Novel Therapeutics for Multiple Sclerosis Designed by Parasitic Worms. Int. J. Mol. Sci. 2017, 18, 2141. [Google Scholar] [CrossRef] [PubMed]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef]
- Morandi, E.; Tarlinton, R.E.; Gran, B. Multiple Sclerosis between Genetics and Infections: Human Endogenous Retroviruses in Monocytes and Macrophages. Front. Immunol. 2015, 6, 647. [Google Scholar] [CrossRef]
- Grandi, N.; Cadeddu, M.; Blomberg, J.; Tramontano, E. Contribution of type W human endogenous retroviruses to the human genome: Characterization of HERV-W proviral insertions and processed pseudogenes. Retrovirology 2016, 13, 67. [Google Scholar] [CrossRef]
- Mechelli, R.; Manzari, C.; Policano, C.; Annese, A.; Picardi, E.; Umeton, R.; Fornasiero, A.; D’Erchia, A.M.; Buscarinu, M.C.; Agliardi, C.; et al. Epstein-Barr virus genetic variants are associated with multiple sclerosis. Neurology 2015, 84, 1362–1368. [Google Scholar] [CrossRef]
- Querol, L.; Clark, P.L.; Bailey, M.A.; Cotsapas, C.; Cross, A.H.; Hafler, D.A.; Kleinstein, S.H.; Lee, J.Y.; Yaari, G.; Willis, S.N.; et al. Protein array-based profiling of CSF identifies RBPJ as an autoantigen in multiple sclerosis. Neurology 2013, 81, 956–963. [Google Scholar] [CrossRef]
- Liu, H.D.; Zheng, H.; Li, M.; Hu, D.S.; Tang, M.; Cao, Y. Upregulated expression of kappa light chain by Epstein-Barr virus encoded latent membrane protein 1 in nasopharyngeal carcinoma cells via NF-kappaB and AP-1 pathways. Cell Signal 2007, 19, 419–427. [Google Scholar] [CrossRef]
- Rinker, J.R., 2nd; Trinkaus, K.; Cross, A.H. Elevated CSF free kappa light chains correlate with disability prognosis in multiple sclerosis. Neurology 2006, 67, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Presslauer, S.; Milosavljevic, D.; Huebl, W.; Aboulenein-Djamshidian, F.; Krugluger, W.; Deisenhammer, F.; Senel, M.; Tumani, H.; Hegen, H. Validation of kappa free light chains as a diagnostic biomarker in multiple sclerosis and clinically isolated syndrome: A multicenter study. Mult. Scler. 2016, 22, 502–510. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarlinton, R.E.; Khaibullin, T.; Granatov, E.; Martynova, E.; Rizvanov, A.; Khaiboullina, S. The Interaction between Viral and Environmental Risk Factors in the Pathogenesis of Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 303. https://doi.org/10.3390/ijms20020303
Tarlinton RE, Khaibullin T, Granatov E, Martynova E, Rizvanov A, Khaiboullina S. The Interaction between Viral and Environmental Risk Factors in the Pathogenesis of Multiple Sclerosis. International Journal of Molecular Sciences. 2019; 20(2):303. https://doi.org/10.3390/ijms20020303
Chicago/Turabian StyleTarlinton, Rachael Eugenie, Timur Khaibullin, Evgenii Granatov, Ekaterina Martynova, Albert Rizvanov, and Svetlana Khaiboullina. 2019. "The Interaction between Viral and Environmental Risk Factors in the Pathogenesis of Multiple Sclerosis" International Journal of Molecular Sciences 20, no. 2: 303. https://doi.org/10.3390/ijms20020303
APA StyleTarlinton, R. E., Khaibullin, T., Granatov, E., Martynova, E., Rizvanov, A., & Khaiboullina, S. (2019). The Interaction between Viral and Environmental Risk Factors in the Pathogenesis of Multiple Sclerosis. International Journal of Molecular Sciences, 20(2), 303. https://doi.org/10.3390/ijms20020303