Kinetics and Predicted Structure of a Novel Xylose Reductase from Chaetomium thermophilum

 and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Protein Production
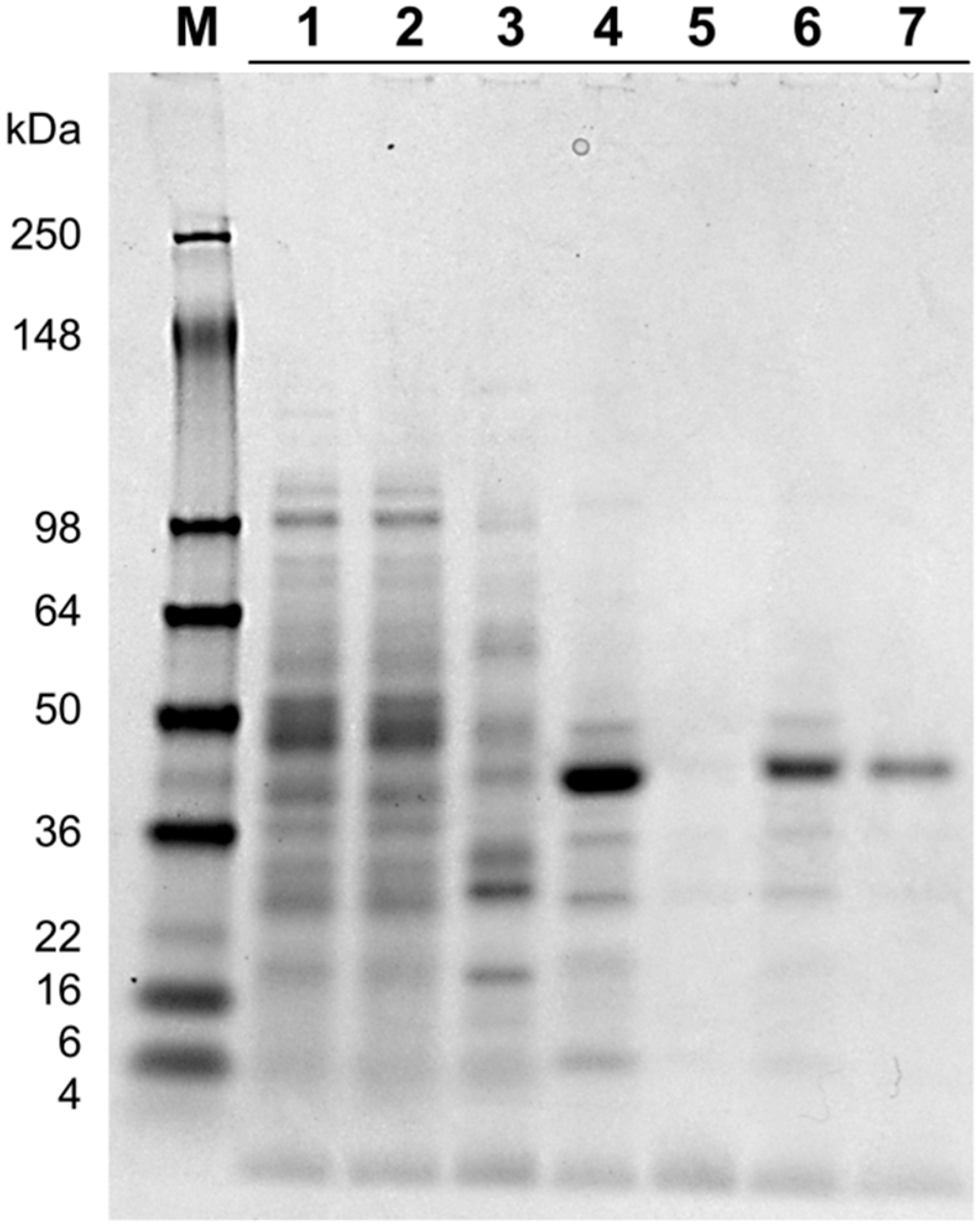
2.2. Protein Purification
2.3. Biochemical Characterization
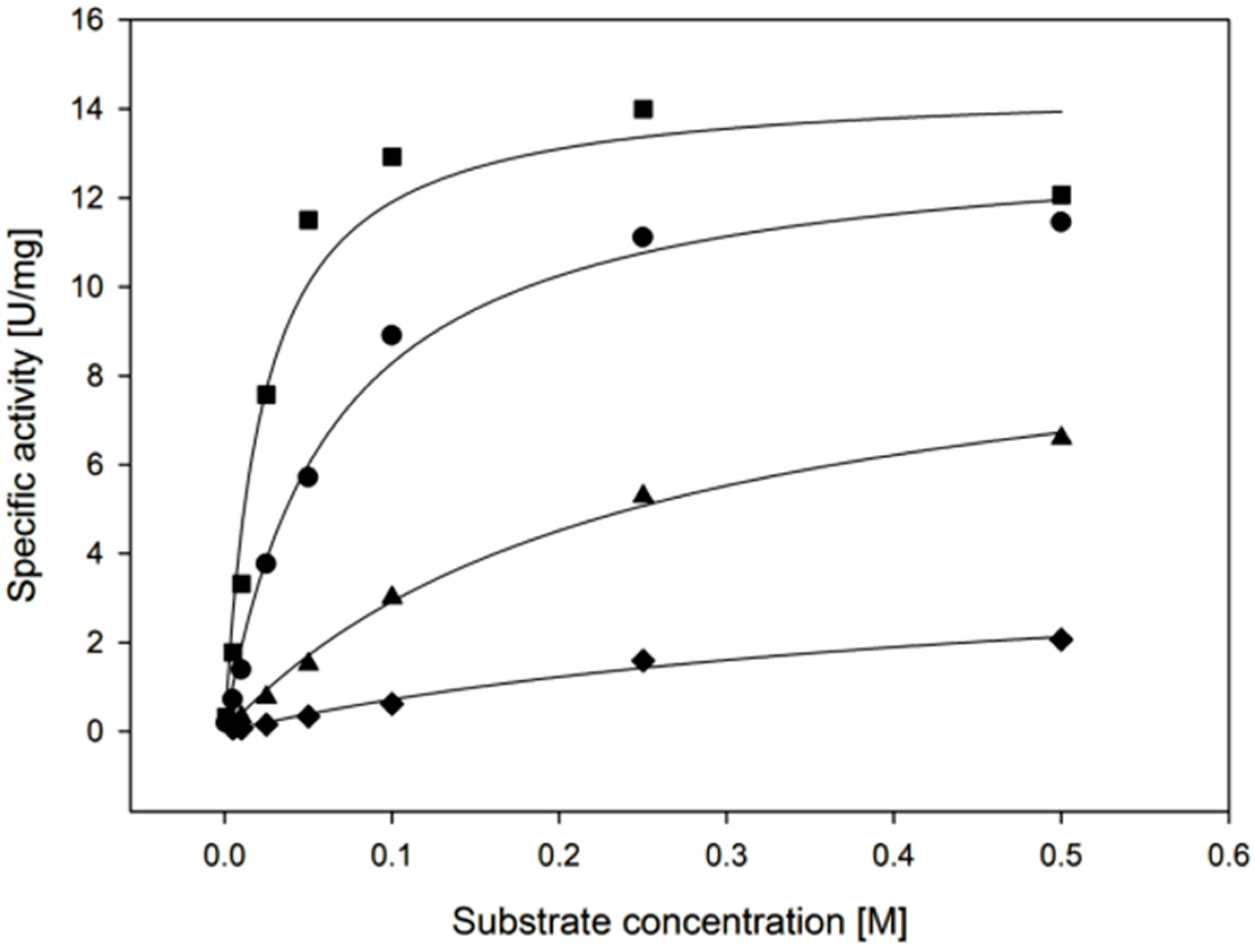
2.3.1. Enzyme Activity
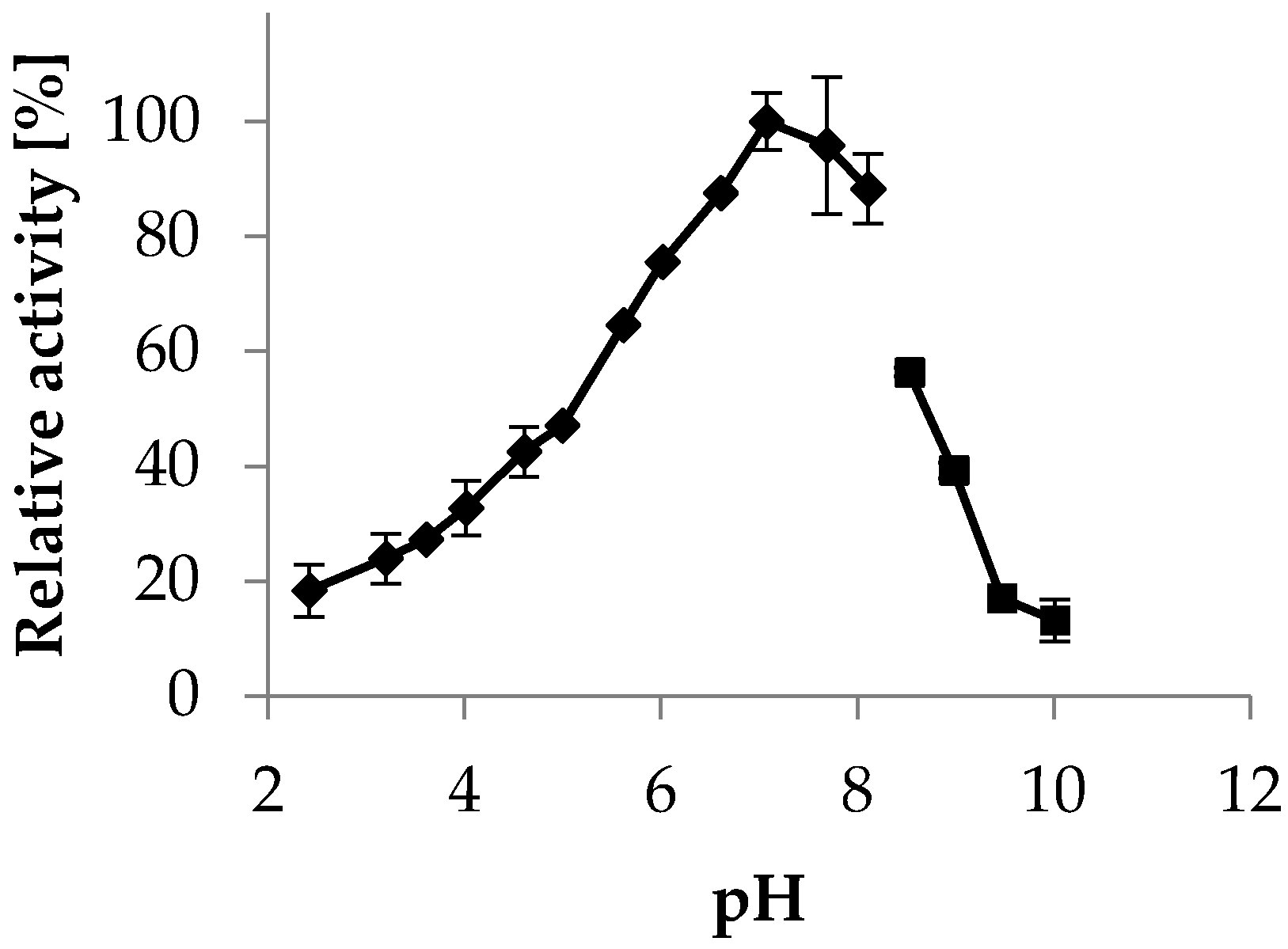
2.3.2. pH Optimum
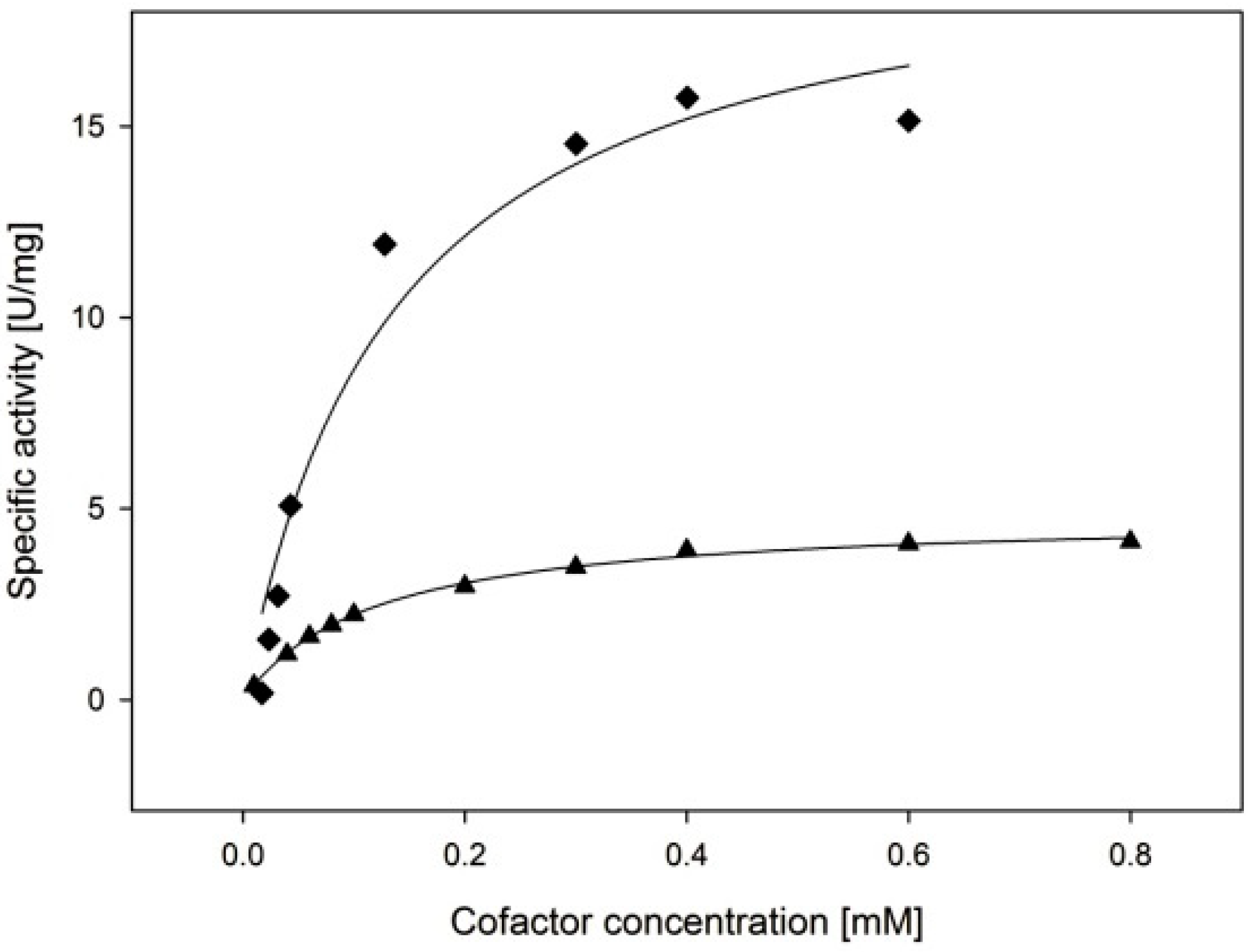
2.3.3. Cofactor Preference
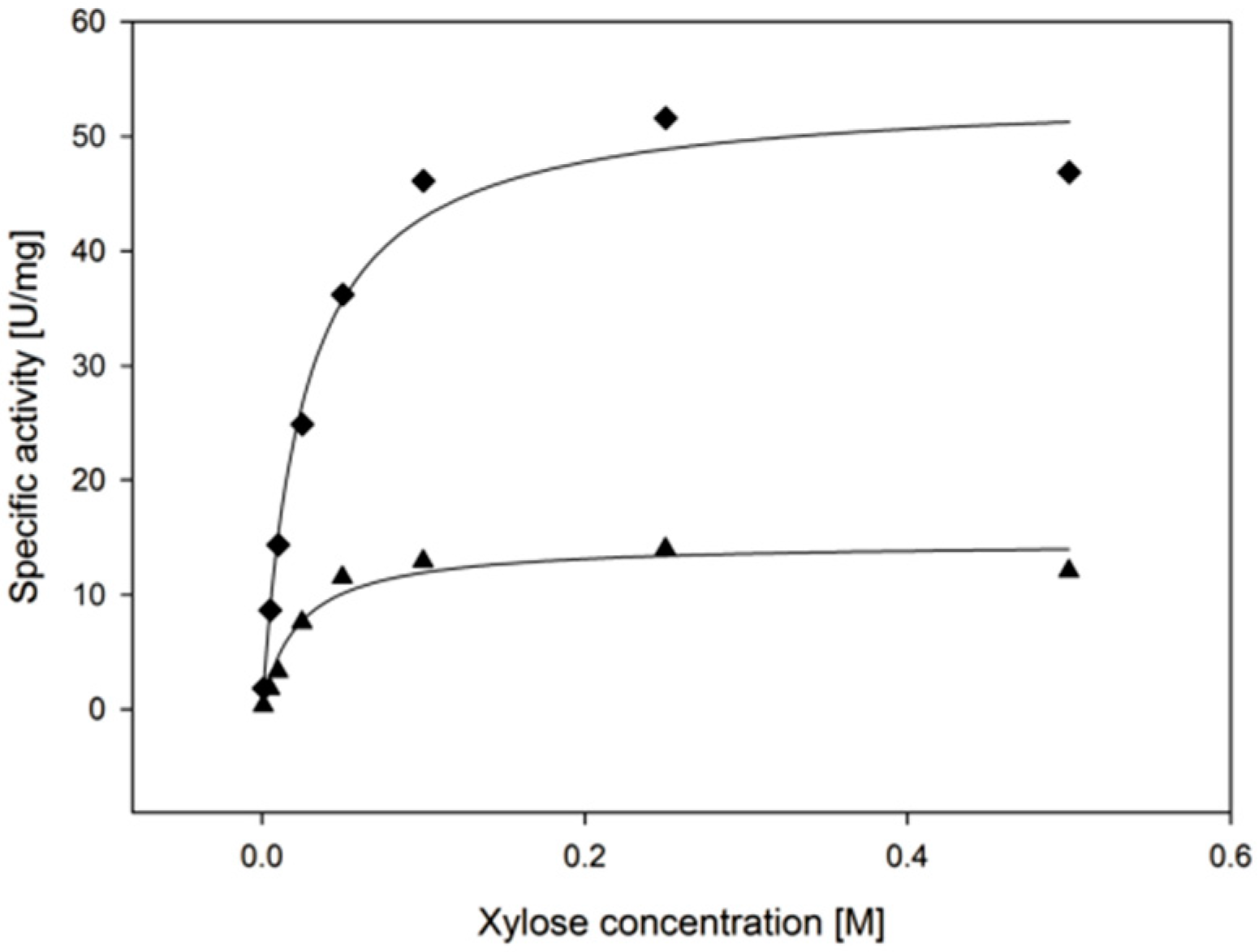
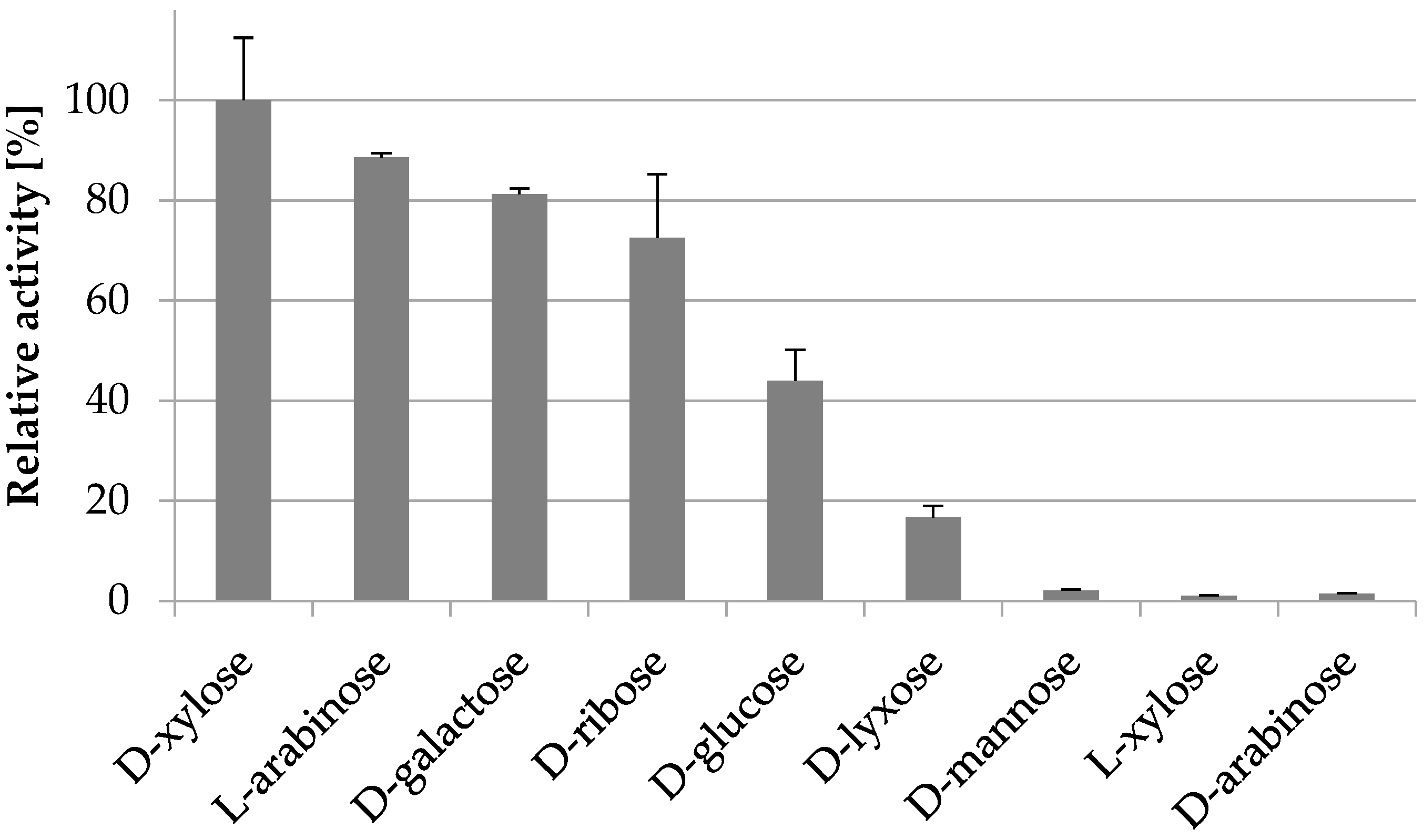
2.3.4. Substrate Specificity
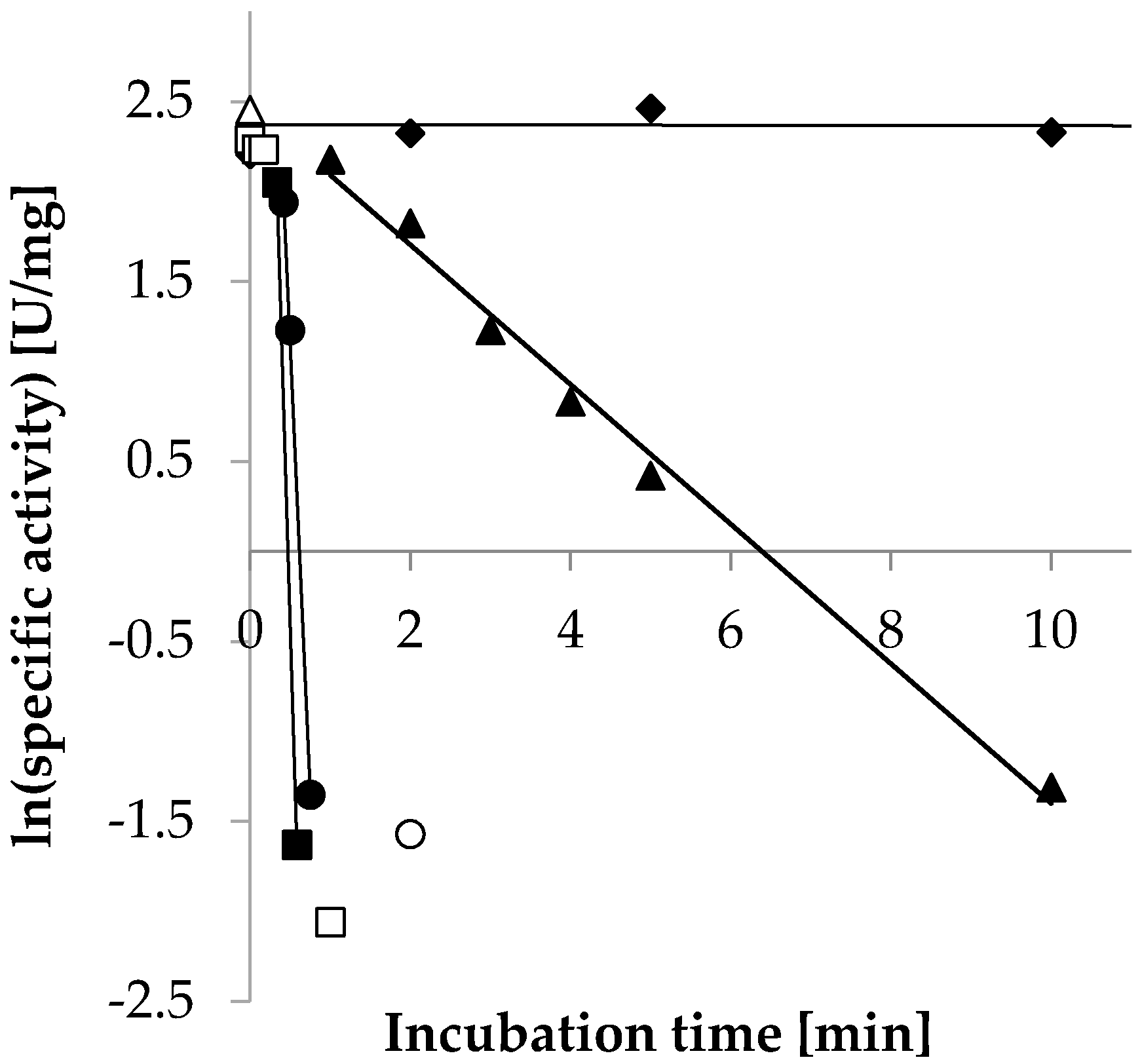
2.3.5. Temperature Stability
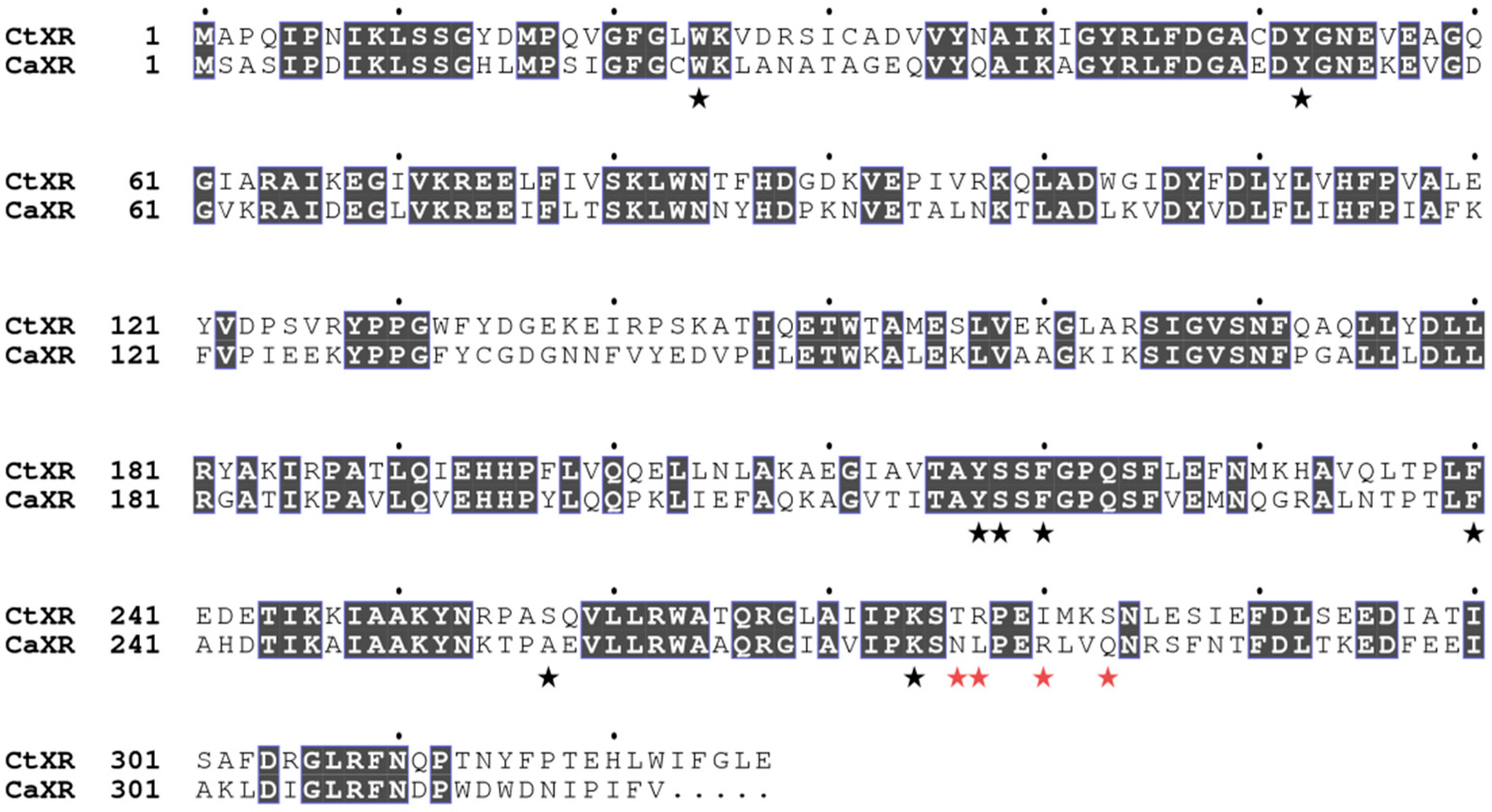
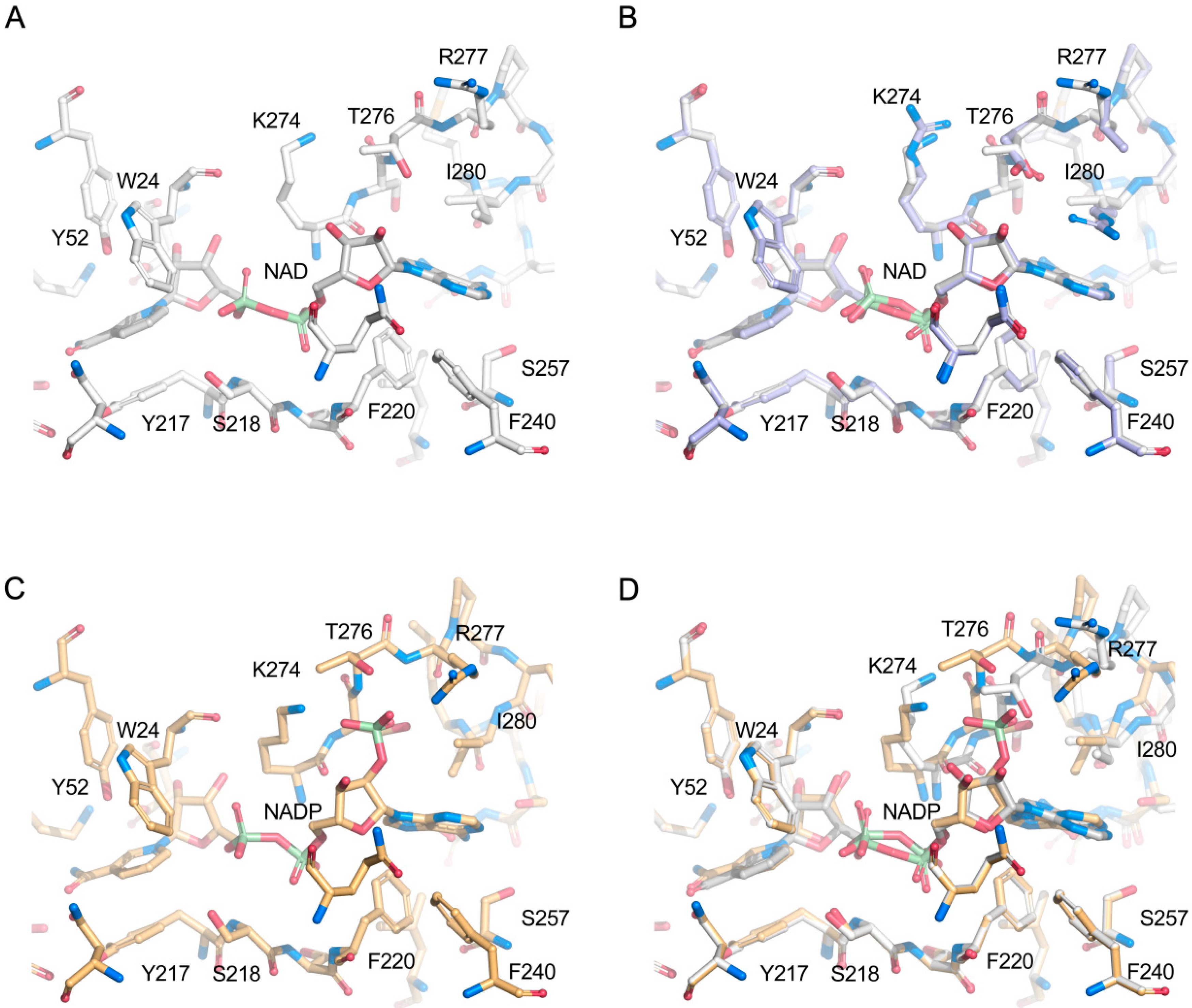
2.4. Structural Analysis
2.5. Comparison with Xylose Reductases in the Literature
4. Materials and Methods
4.1. Materials and Reagents
4.2. Construction of the Expression Plasmid
4.3. Protein Production
4.4. Protein Purification
4.5. Data Analysis
4.6. Protein Quantification
4.7. Biochemical Characterization
4.7.1. Enzyme Activity
4.7.2. pH Optimum
4.7.3. Cofactor Preference
4.7.4. Substrate Specificity
4.7.5. Temperature Stability
4.8. Homology Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| XR | xylose reductase |
| His6 | hexahistidine |
| NADPH | nicotinamide adenine dinucleotide phosphate (reduced) |
| NADH | nicotinamide adenine dinucleotide (reduced) |
| PDB | Protein Data Bank |
| EC | Enzyme Commission (number) |
| NAD+ | nicotinamide adenine dinucleotide (oxidized) |
| CtXR | xylose reductase from Chaetomium thermophilum |
| DCW | dry cell weight (g/L) |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| IMAC | immobilized metal affinity chromatography |
| SEC | size exclusion chromatography |
| Vmax | maximum enzyme activity (molNAD(P)H·s−1·mgXR−1) |
| kcat | turnover number (molNAD(P)H·molXR−1·s−1) |
| KM | substrate affinity (Msubstrate) |
| kcat/KM | catalytic efficiency (molNAD(P)H·molXR−1·MSubstrate.−1·s−1) |
| SE | standard error |
| CaXR | xylose reductase from Candida tenuis |
| GMQE | global model quality estimation |
| SsADH | aldehyde reductase from Sus scrofa |
| CV | column volume (mL) |
| NAD(H) | nicotinamide adenine dinucleotide |
| NADP(H) | nicotinamide adenine dinucleotide phosphate |
References
- Bock, T.; Chen, W.-H.; Ori, A.; Malik, N.; Silva-Martin, N.; Huerta-Cepas, J.; Powell, S.T.; Kastritis, P.L.; Smyshlyaev, G.; Vonkova, I.; et al. An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 2014, 42, 13525–13533. [Google Scholar] [CrossRef] [PubMed]
- Amlacher, S.; Sarges, P.; Flemming, D.; van Noort, V.; Kunze, R.; Devos, D.P.; Arumugam, M.; Bork, P.; Hurt, E. Insight into Structure and Assembly of the Nuclear Pore Complex by Utilizing the Genome of a Eukaryotic Thermophile. Cell 2011, 146, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Stetter, K.O. Hyperthermophilic Microorganisms; Springer: Berlin, Germany, 2002. [Google Scholar]
- Häcker, B.; Habenicht, A.; Kiess, M.; Mattes, R. Xylose Utilisation: Cloning and Characterisation of the Xylose Reductase from Candida tenuis. Biol. Chem. 2005, 380, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Mayr, P.; Brüggler, K.; Kulbe, K.D.; Nidetzky, B. D-Xylose metabolism by Candida intermedia: Isolation and characterisation of two forms of aldose reductase with different coenzyme specificities. J. Chromatogr. B 2000, 737, 195–202. [Google Scholar] [CrossRef]
- Neuhauser, W.; Haltrich, D.; Kulbe, K.D.; Nidetzky, B. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis: Isolation, characterization and biochemical properties of the enzyme. Biochem. J. 1997, 326, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Verduyn, C.; Kleef, R.V.; Frank, J.; Schreuder, H.; Dijken, J.P.V.; Scheffers, W.A. Properties of the NAD(P)H-dependent xylose reductase from the xylose-fermenting yeast Pichia stipitis. Biochem. J. 1985, 226, 669–677. [Google Scholar] [CrossRef]
- Lee, J.-K.; Koo, B.-S.; Kim, S.-Y. Cloning and Characterization of the xyl1 Gene, Encoding an NADH-Preferring Xylose Reductase from Candida parapsilosis, and Its Functional Expression in Candida tropicalis. Appl. Environ. Microbiol. 2003, 69, 6179–6188. [Google Scholar] [CrossRef]
- Bengtsson, O.; Hahn-Hägerdal, B.; Gorwa-Grauslund, M.F. Xylose reductase from Pichia stipitis with altered coenzyme preference improves ethanolic xylose fermentation by recombinant Saccharomyces cerevisiae. Biotechnol. Biofuels 2009, 2, 9. [Google Scholar] [CrossRef]
- Fernandes, S.; Tuohy, M.G.; Murray, P.G. Xylose reductase from the thermophilic fungus Talaromyces emersonii: Cloning and heterologous expression of the native gene (Texr) and a double mutant (TexrK271R+N273D) with altered coenzyme specificity. J. Biosci. 2009, 34, 881–890. [Google Scholar] [CrossRef]
- Krahulec, S.; Klimacek, M.; Nidetzky, B. Analysis and prediction of the physiological effects of altered coenzyme specificity in xylose reductase and xylitol dehydrogenase during xylose fermentation by Saccharomyces cerevisiae. J. Biotechnol. 2012, 158, 192–202. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kodaki, T.; Park, Y.-C.; Seo, J.-H. Effects of NADH-preferring xylose reductase expression on ethanol production from xylose in xylose-metabolizing recombinant Saccharomyces cerevisiae. J. Biotechnol. 2012, 158, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Petschacher, B.; Nidetzky, B. Altering the coenzyme preference of xylose reductase to favor utilization of NADH enhances ethanol yield from xylose in a metabolically engineered strain of Saccharomyces cerevisiae. Microb. Cell Fact. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Abu Saleh, A.; Pack, S.P.; Annaluru, N.; Kodaki, T.; Makino, K. Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein-engineered NADH-preferring xylose reductase from Pichia stipitis. Microbiology 2007, 153, 3044–3054. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.-K.; Du, H.-L.; Wang, J.-F.; Wei, D.-Q.; Wang, X.-N.; Li, Y.-X.; Lin, Y. Reversal of coenzyme specificity and improvement of catalytic efficiency of Pichia stipitis xylose reductase by rational site-directed mutagenesis. Biotechnol. Lett. 2009, 31, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Sanli, G.; Dudley, J.I.; Blaber, M. Structural biology of the aldo-keto reductase family of enzymes. Cell Biochem. Biophys. 2003, 38, 79–101. [Google Scholar] [CrossRef]
- Kavanagh, K.L.; Klimacek, M.; Nidetzky, B.; Wilson, D.K. The Structure of Apo and Holo Forms of Xylose Reductase, a Dimeric Aldo-Keto Reductase from Candida tenuis. Biochemistry 2002, 41, 8785–8795. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.; Jin, Y.-S. Production of fuels and chemicals from xylose by engineered Saccharomyces cerevisiae: A review and perspective. Microb. Cell Fact. 2017, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mishra, P. Microbial Pentose Utilization: Current Applications in Biotechnology; Elsevier: Amsterdam, The Netherlands, 1995; ISBN 978-0-08-054493-9. [Google Scholar]
- Edelstein, S.; Smith, K.; Worthington, A.; Gillis, N.; Bruen, D.; Kang, S.H.; Ho, W.L.; Gilpin, K.; Ackerman, J.; Guiducci, G. Comparisons of Six New Artificial Sweetener Gradation Ratios with Sucrose in Conventional-Method Cupcakes Resulting in Best Percentage Substitution Ratios. J. Culin. Sci. Technol. 2007, 5, 61–74. [Google Scholar] [CrossRef]
- De Albuquerque, T.L.; da Silva, I.J., Jr.; de Macedo, G.R.; Rocha, M.V.P. Biotechnological production of xylitol from lignocellulosic wastes: A review. Process Biochem. 2014, 49, 1779–1789. [Google Scholar] [CrossRef]
- Dasgupta, D.; Bandhu, S.; Adhikari, D.K.; Ghosh, D. Challenges and prospects of xylitol production with whole cell bio-catalysis: A review. Microbiol. Res. 2017, 197, 9–21. [Google Scholar] [CrossRef]
- Grand View Research, Inc. Xylitol Market Size Worth $1.37 Billion By 2025 | Growth Rate: 6.6%; Grand View Research, Inc.: San Francisco, CA, USA, 2017; Available online: https://www.grandviewresearch.com/press-release/global-xylitol-market (accessed on 28 August 2018).
- Woodyer, R.; Simurdiak, M.; Van Der Donk, W.A.; Zhao, H. Heterologous expression, purification, and characterization of a highly active xylose reductase from Neurospora crassa. Appl. Environ. Microbiol. 2005, 71, 1642–1647. [Google Scholar] [CrossRef]
- Kavanagh, K.L.; Klimacek, M.; Nidetzky, B.; Wilson, D.K. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochem. J. 2003, 373, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Hakulinen, N.; Turunen, O.; Jänis, J.; Leisola, M.; Rouvinen, J. Three-dimensional structures of thermophilic β-1,4-xylanases from Chaetomium thermophilum and Nonomuraea flexuosa. Eur. J. Biochem. 2003, 270, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Petschacher, B.; Leitgeb, S.; Kavanagh, K.L.; Wilson, D.K.; Nidetzky, B. The coenzyme specificity of Candida tenuis xylose reductase (AKR2B5) explored by site-directed mutagenesis and X-ray crystallography. Biochem. J. 2005, 385, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qiao, D.; Xu, H.; Liao, C.; Li, S.; Cao, Y. Cloning, expression, and characterization of xylose reductase with higher activity from Candida tropicalis. J. Microbiol. 2009, 47, 351–357. [Google Scholar] [CrossRef]
- Yokoyama, S.-I.; Suzuki, T.; Kawai, K.; Horitsu, H.; Takamizawa, K. Purification, characterization and structure analysis of NADPH-dependent d-xylose reductases from Candida tropicalis. J. Ferment. Bioeng. 1995, 79, 217–223. [Google Scholar] [CrossRef]
- DeLisa, M.P.; Li, J.; Rao, G.; Weigand, W.A.; Bentley, W.E. Monitoring GFP-operon fusion protein expression during high cell density cultivation of Escherichia coli using an on-line optical sensor. Biotechnol. Bioeng. 1999, 65, 54–64. [Google Scholar] [CrossRef]
- Wu, J.T.; Wu, L.H.; Knight, J.A. Stability of NADPH: Effect of various factors on the kinetics of degradation. Clin. Chem. 1986, 32, 314–319. [Google Scholar]
- McIlvaine, T.C. A Buffer Solution for Colorimetric Comparison. J. Biol. Chem. 1921, 49, 183–186. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Hyndman, D.; Green, N.; Li, X.; Korithoski, B.; Jia, Z.; Flynn, T.G. Crystal structure of an aldehyde reductase Y50F mutant-NADP complex and its implications for substrate binding. Proteins Struct. Funct. Bioinform. 2001, 44, 12–19. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Protein | Specific Activity | Total Activity | Recovery | Purification Factor | |
|---|---|---|---|---|---|
| (mg) | (U/mg) | (U) | (%) | ( ) | |
| Crude extract | 4296 | 0.08 | 359 | 100 | 1 |
| IMAC Flow through | 4158 | 0.09 | 392 | 109 | 1.1 |
| IMAC Eluate 1 (pre elution peak) | 11.85 | 0.28 | 3.3 | 0.9 | 3.4 |
| IMAC Eluate 2 (post elution peak) | 3.96 | 0.12 | 0.5 | 0.1 | 1.4 |
| IMAC Elution peak | 55.7 | 1.49 | 83.1 | 23.1 (23.1) | 17.8 (17.8) |
| Spin filter Flow through | 25.1 | 2.12 | 53.1 | 14.8 | 25.3 |
| Spin filter retentate | 34.1 | 0.76 | 26.0 | 7.2 (31.3) | 9.1 (0.5) |
| SEC Pool 1 (discarded fraction) | 13.4 | 0.76 | 10.1 | 2.8 | 9.0 |
| SEC Pool 2 (discarded fraction) | 5.20 | 0.86 | 4.5 | 1.2 | 10.3 |
| SEC Peak (collected fraction) | 4.51 | 2.13 | 9.6 | 2.7 (36.9) | 25.5 (2.8) |
| Temperature | kcat (s−1) | KM, (Xylose) (mM) | kcat/KM, (Xylose) (mM−1·min−1) |
|---|---|---|---|
| 30 °C | 11.4 ± 1.1 | 22.3 ± 6.1 | 31 ± 9 |
| 55 °C | 35.2 ± 1.4 | 25.4 ± 4.0 | 83 ± 14 |
| Cofactor | kcat (s−1) | KM, (Cofactor) (µM) | kcat/KM, (Cofactor) (µM−1·min−1) |
|---|---|---|---|
| NADPH | 11.4 ± 1.1 | 135 ± 44 | 5.1 ± 1.7 |
| NADH | 3.2 ± 0.1 | 119 ± 6 | 1.6 ± 0.1 |
| Substrate | kcat (s−1) | KM, (Substrate) (mM) | kcat/KM, (Substrate) (mM−1·min−1) |
|---|---|---|---|
| d-xylose | 9.2 ± 0.6 | 22.3 ± 6.1 | 26 ± 7 |
| l-arabinose | 8.8 ± 0.4 | 62.5 ± 7.7 | 8.4 ± 1.1 |
| d-galactose | 6.5 ± 0.4 | 241 ± 29 | 1.6 ± 0.2 |
| d-glucose | 2.7 ± 0.5 | 471 ± 141 | 0.3 ± 0.1 |
| Temperature | Decay Rate (λ) (s−1) | Half-Life (T1/2) |
|---|---|---|
| ◆ 30 °C | n.a. | n.a. |
| ▲ 55 °C | −6.5 × 10−3 | 1.8 min |
| ● 65 °C | −0.17 | 4.2 s |
| ■ 75 °C | −0.25 | 2.8 s |
| Organism and Source | Cofactor | kcat (s−1) | KM, (Xylose) (mM) | kcat/KM, (Xylose) (mM−1·min−1) | KM, (Cofactor) (µM) | kcat/KM, (Cofactor) (µM−1·min−1) | Assay Conditions | Optimum | Temp. Stability | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | pH | Temp. (°C) | pH | ||||||||
| Chaetomium thermophilum (this work) | NADPH | 35.2 | 25.4 | 83 | 55 | 6.5 | 55 | 6.5 | No loss of activity after 3 h at 30 °C T1/2 = 1.8 min (55 °C) | ||
| NADH | 3.2 | 119 | 1.6 | 30 | |||||||
| Neurospora crassa [24] | NADPH | 60 | 34 | 106 | 1.8 | 2000 | 25 | 6.3 | 45–55 | 5.5 | T1/2 = 49 min (40 °C) |
| NADH | 5.2 | 37 | 8.4 | 16 | 19.5 | ||||||
| Candida tenuis [4,6] | NADPH | 21.7 | 72 | 18.1 | 4.8 | 271 | 25 | 7.0 | 50 | 6.0 | Start of decrease at 30-35 °C |
| NADH | 18.2 | 87 | 12.6 | 25.4 | 43 | ||||||
| Candida tropicalis [30] | NADPH | 49 | 30 | 99 | 18 | 329 | RT | 7.0 | - | 6.0 | Complete loss after 1 h at 60 °C |
| Candida intermedia [5] | NADH | 16.5 | 30 | 31.8 | 10 | 101 | 25 | 7.0 | - | - | - |
| Pichia stipitis [7] | NADPH | 25.2 | 42 | 36 | 9 | 167 | 30 | 6.0 | - | 6.0 | - |
| Candida parapsilosis [8] | NADPH | 4.6 | 244 | 1.14 | 36.5 | 7.6 | 37 | 6.0 | - | 6.0 | T1/2 = 4.5 h (45 °C); 2 min (50 °C) |
| NADH | 49 | 32 | 87.6 | 3.3 | 939 | ||||||
| Candida tenuis [28] | NADPH | 13 | 96 | 8.1 | 3 | 260 | 25 | 7.0 | - | - | - |
| NADH | 11 | 142 | 4.6 | 38 | 17 | ||||||
| Candida tropicalis [29] | NADPH | 240 | 31.5 | 457 | 45.5 | 316 | 45 | 5.5 | 45 | 5.5 | T1/2 = 15 min (60 °C) |
| NADH | - | - | - | 162 | - | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quehenberger, J.; Reichenbach, T.; Baumann, N.; Rettenbacher, L.; Divne, C.; Spadiut, O. Kinetics and Predicted Structure of a Novel Xylose Reductase from Chaetomium thermophilum. Int. J. Mol. Sci. 2019, 20, 185. https://doi.org/10.3390/ijms20010185
Quehenberger J, Reichenbach T, Baumann N, Rettenbacher L, Divne C, Spadiut O. Kinetics and Predicted Structure of a Novel Xylose Reductase from Chaetomium thermophilum. International Journal of Molecular Sciences. 2019; 20(1):185. https://doi.org/10.3390/ijms20010185
Chicago/Turabian StyleQuehenberger, Julian, Tom Reichenbach, Niklas Baumann, Lukas Rettenbacher, Christina Divne, and Oliver Spadiut. 2019. "Kinetics and Predicted Structure of a Novel Xylose Reductase from Chaetomium thermophilum" International Journal of Molecular Sciences 20, no. 1: 185. https://doi.org/10.3390/ijms20010185
APA StyleQuehenberger, J., Reichenbach, T., Baumann, N., Rettenbacher, L., Divne, C., & Spadiut, O. (2019). Kinetics and Predicted Structure of a Novel Xylose Reductase from Chaetomium thermophilum. International Journal of Molecular Sciences, 20(1), 185. https://doi.org/10.3390/ijms20010185