A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV)

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
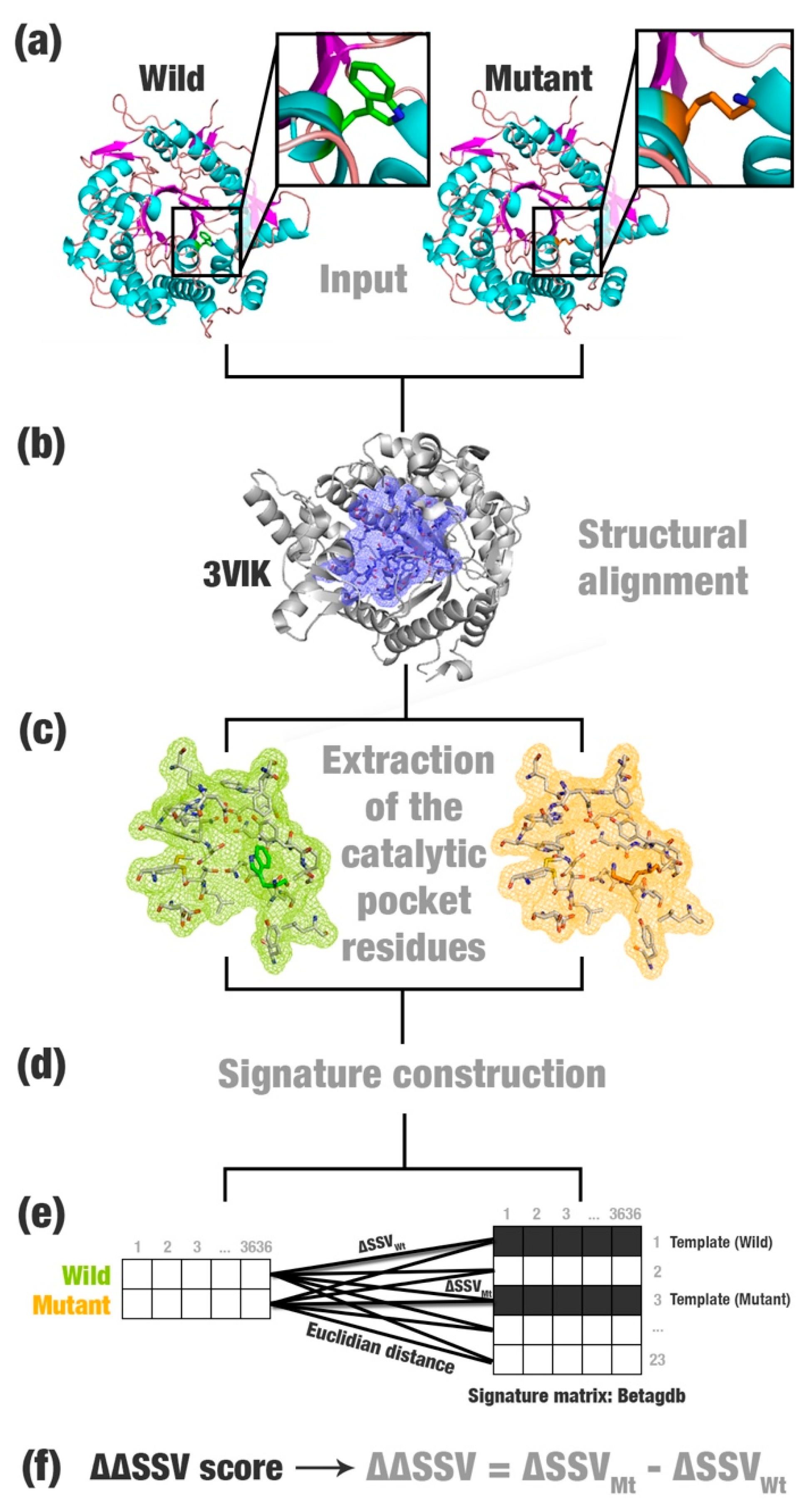
2.1. SSV Definition
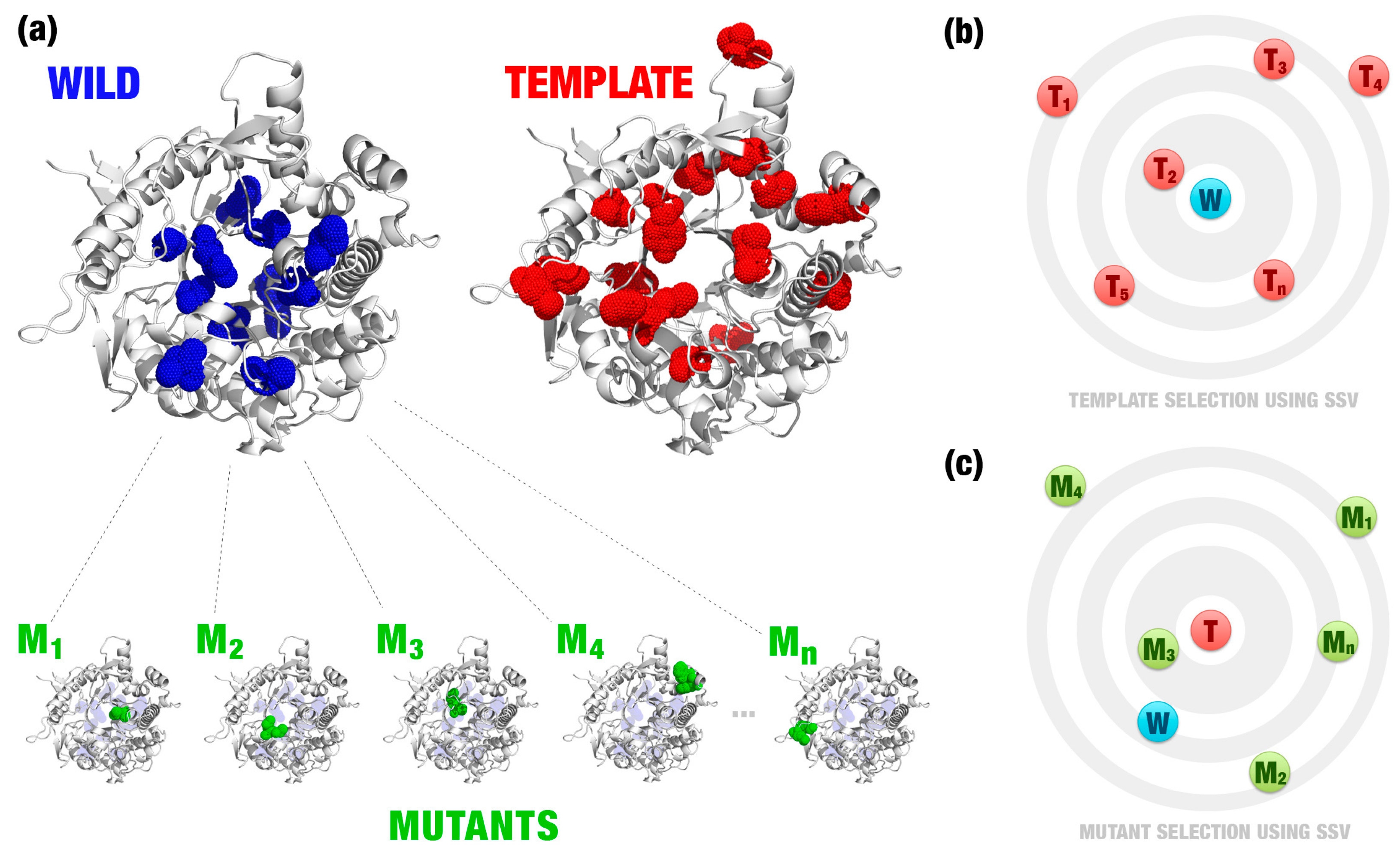
- Most relevant residues’ extraction: For wild, mutated, and templates’ structures the most relevant residues are extracted and saved in a new Protein Data Bank (PDB) file (Figure 1a–c). This selection depends on the application and can be modified according to users’ needs. This step is optional.
- Structural signature construction: For every PDB file, we compute a vector with the cumulative distribution of the pairwise distances among all pairs of atoms and their physicochemical proprieties (aCSM algorithm) (Figure 1d).
- Template definition: A template definition depends on a high-curated database of enzymes with beneficial characteristics. This database should be manually and previously defined. We selected as a template, proteins with the closest signature to wild and mutant proteins analyzed (Figure 1e).
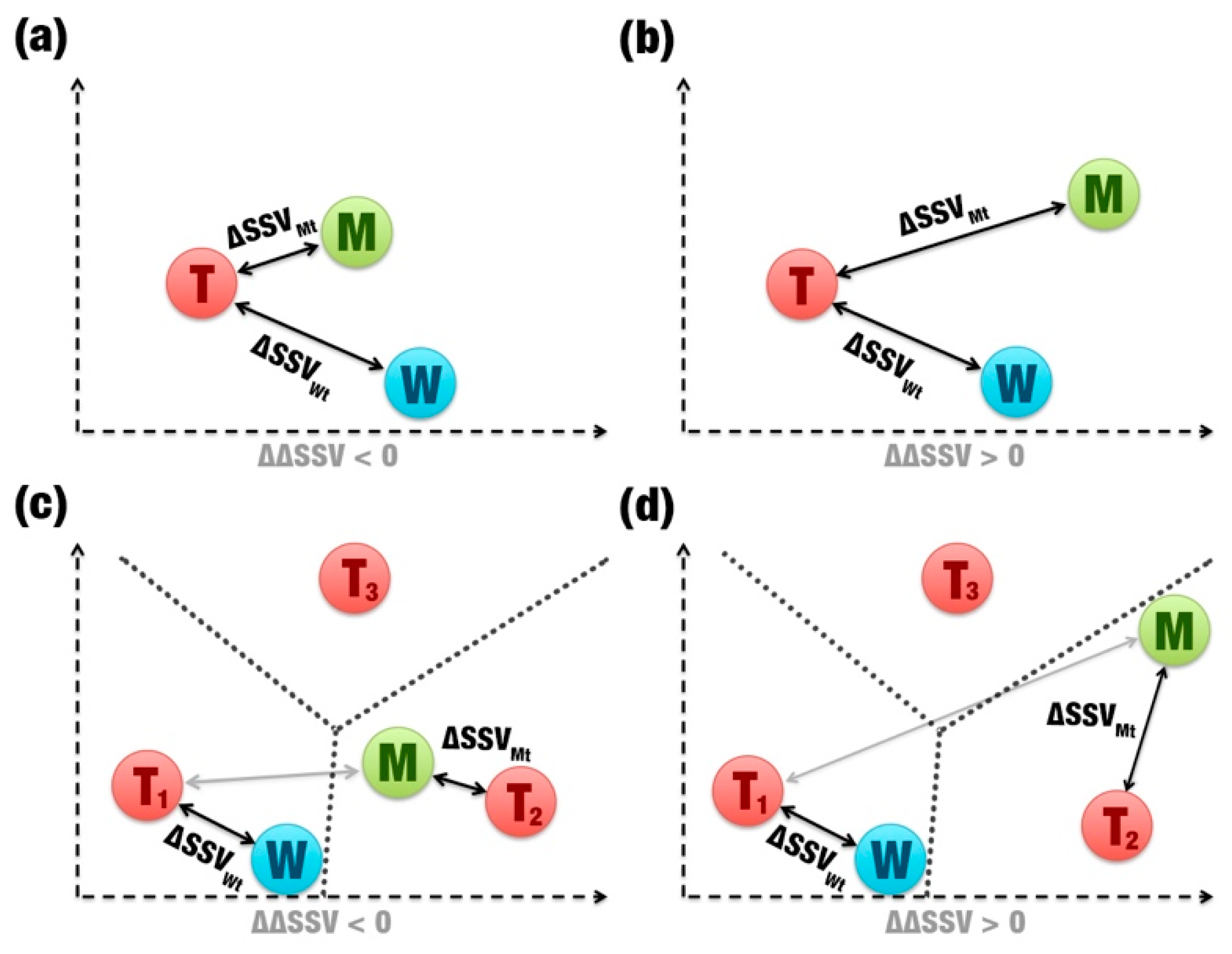
- Comparison between signatures: A distance matrix among all signatures is constructed (a similar matrix is used to define the template). The Euclidean distance between two signatures is called signature variation (ΔSSV). The Euclidean distance between signatures of a wild enzyme and its template is called (ΔSSVWt). The Euclidean distance between signatures of a mutant enzyme and its template is called (ΔSSVMt). The difference between both values is the ΔΔSSV score. If the ΔΔSSV score is lower than zero, the mutant’s signature is more alike to the template signature than to the wild’s signature, suggesting that the mutation is beneficial. If the ΔΔSSV score is higher than zero, the mutant’s signature is more distant from the template signature than from wild’s signature, suggesting that the mutation is not beneficial (Figure 1f).
2.2. Case Study 1: Evaluating Mutations for β-Glucosidase Collected from the Literature
2.2.1 Data Collection and Manual Classification of Mutation Effects
2.2.2. Predicting the Impact of Mutations
2.2.3. Comparison with Other Methods
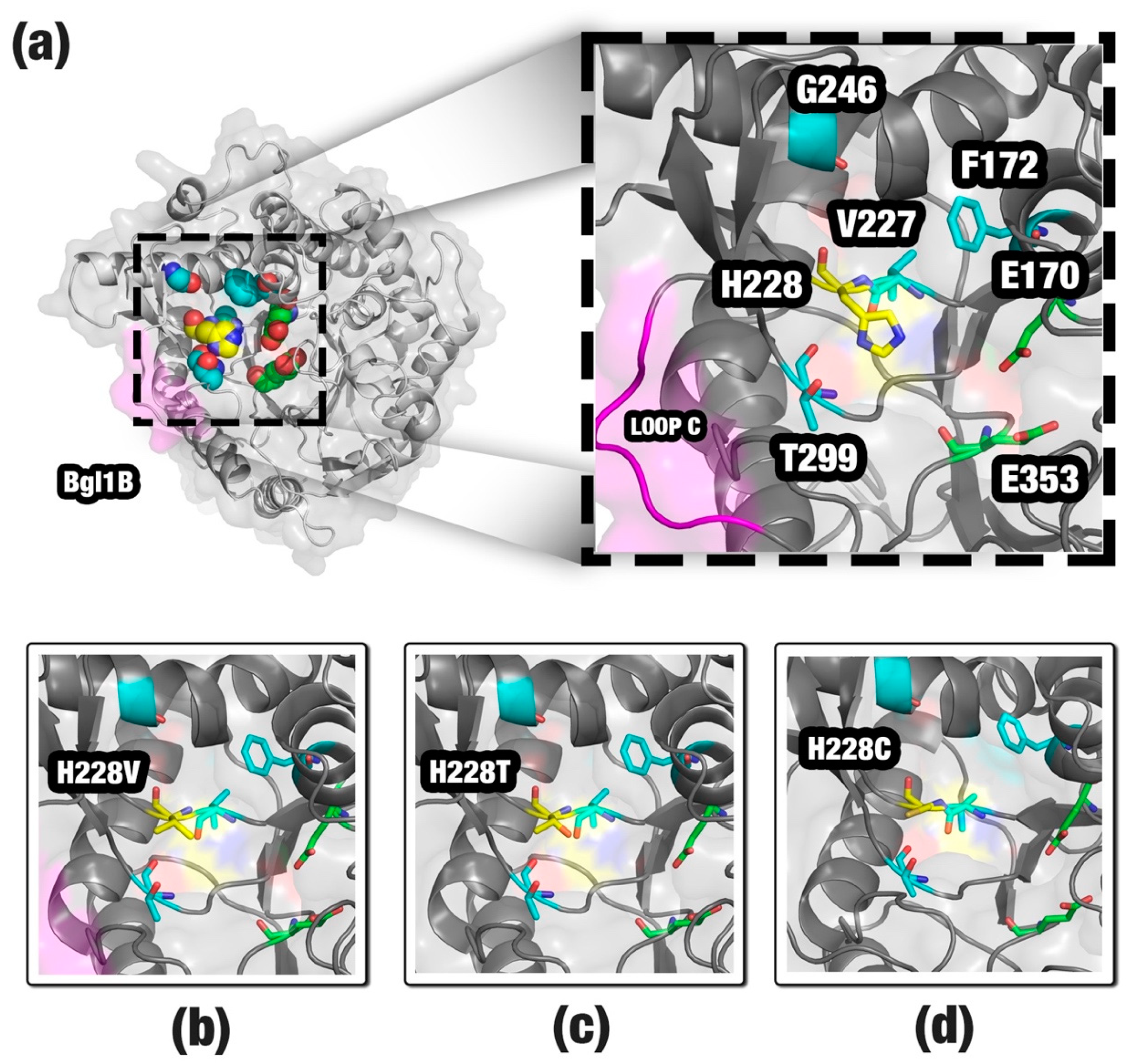
2.3. Case Study 2: Proposing Mutations for a Non-Tolerant β-Glucosidase
2.4. Case Study 3: Comparing to BioGPS Descriptors
3. Discussion
3.1. Improving the Activity of a Non-Tolerant β-Glucosidase
3.2. Evaluating Mutations in CaLB
3.3. Important Issues before Using SSV
4. Materials and Methods
4.1. Method Description
4.1.1 Extraction of the Catalytic Pocket
4.1.2. Structural Signature Construction
4.1.3. Template Definition
4.1.4. Comparison between Signatures
4.2. Case Study 1
4.3. Case Study 2
4.4. Case Study 3
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CaLB | Candida antarctica lipase B |
| epPCR | Error-prone PCR |
| GH1 | Glycoside hydrolase family 1 |
| IF | Improvement Factor |
| SSV | Structural Signature Variation |
| SVM | Support Vector Machine |
References
- Chaudhary, N.; Gupta, A.; Gupta, S.; Sharma, V.K. BioFuelDB: A database and prediction server of enzymes involved in biofuels production. PeerJ 2017, 5, e3497. [Google Scholar] [CrossRef] [PubMed]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. Characterization of protein hydrolysates from blue whiting (Micromesistius poutassou) and their application in beverage fortification. Food Chem. 2018, 245, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, Z.; Ren, G.; Kong, W.; Li, L.; Xie, W.; Liu, Y. Engineering a novel glucose-tolerant β-glucosidase as supplementation to enhance the hydrolysis of sugarcane bagasse at high glucose concentration. Biotechnol. Biofuels 2015, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Cirino, P.C.; Mayer, K.M.; Umeno, D. Generating mutant libraries using error-prone PCR. Methods Mol. Biol. 2003, 231, 3–9. [Google Scholar] [PubMed]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 2007, 25, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Berland, M.; Offmann, B.; André, I.; Remaud-Siméon, M.; Charton, P. A web-based tool for rational screening of mutants libraries using ProSAR. Protein Eng. Des. Sel. 2014, 27, 375–381. [Google Scholar] [CrossRef]
- Christ, C.D.; Fox, T. Accuracy assessment and automation of free energy calculations for drug design. J. Chem. Inf. Model. 2014, 54, 108–120. [Google Scholar] [CrossRef]
- Pires, D.E.; de Melo-Minardi, R.C.; dos Santos, M.A.; da Silveira, C.H.; Santoro, M.M.; Meira, W. Cutoff Scanning Matrix (CSM): Structural classification and function prediction by protein inter-residue distance patterns. BMC Genomics 2011, 12, S12. [Google Scholar] [CrossRef]
- Pires, D.E.V. CSM: Uma assinatura para grafos biológicos baseada em padrões de distâncias. Ph.D. Thesis, Universidade Federal de Minas Gerais (UFMG), Belo Horizonte-MG, Brazil, 2012. [Google Scholar]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinform. Oxf. Engl. 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. mCSM-lig: Quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci. Rep. 2016, 6, 29575. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B. mCSM-AB: A web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 2016, 44, W469–W473. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; de Melo-Minardi, R.C.; da Silveira, C.H.; Campos, F.F.; Meira, W. aCSM: Noise-free graph-based signatures to large-scale receptor-based ligand prediction. Bioinform. Oxf. Engl. 2013, 29, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, V.; Siragusa, L.; Ebert, C.; Baroni, M.; Foscato, M.; Cruciani, G.; Gardossi, L. BioGPS descriptors for rational engineering of enzyme promiscuity and structure based bioinformatic analysis. PLoS ONE 2014, 9, e109354. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.R.K.; Esen, A. β-Glucosidases. Cell. Mol. Life Sci. 2010, 67, 3389–3405. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Du, L.; Wei, Y.; Hu, Y.; Huang, R. Expression and characterization of a novel highly glucose-tolerant β-glucosidase from a soil metagenome. Acta Biochim. Biophys. Sin. 2013, 45, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Béguin, P.; Aubert, J.P. The biological degradation of cellulose. FEMS Microbiol. Rev. 1994, 13, 25–58. [Google Scholar] [CrossRef]
- Murphy, L.; Bohlin, C.; Baumann, M.J.; Olsen, S.N.; Sørensen, T.H.; Anderson, L.; Borch, K.; Westh, P. Product inhibition of five Hypocrea jecorina cellulases. Enzyme Microb. Technol. 2013, 52, 163–169. [Google Scholar] [CrossRef]
- Chamoli, S.; Kumar, P.; Navani, N.K.; Verma, A.K. Secretory expression, characterization and docking study of glucose-tolerant β-glucosidase from B. subtilis. Int. J. Biol. Macromol. 2016, 85, 425–433. [Google Scholar] [CrossRef]
- Kadam, S.K.; Demain, A.L. Addition of cloned beta-glucosidase enhances the degradation of crystalline cellulose by the Clostridium thermocellum cellulose complex. Biochem. Biophys. Res. Commun. 1989, 161, 706–711. [Google Scholar] [CrossRef]
- Watanabe, T.; Sato, T.; Yoshioka, S.; Koshijima, T.; Kuwahara, M. Purification and properties of Aspergillus niger beta-glucosidase. Eur. J. Biochem. FEBS 1992, 209, 651–659. [Google Scholar] [CrossRef]
- Zhao, L.; Pang, Q.; Xie, J.; Pei, J.; Wang, F.; Fan, S. Enzymatic properties of Thermoanaerobacterium thermosaccharolyticum β-glucosidase fused to Clostridium cellulovorans cellulose binding domain and its application in hydrolysis of microcrystalline cellulose. BMC Biotechnol. 2013, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, Y.; Chemardin, P.; Arnaud, A.; Galzy, P. Purification and characterization of an intracellular β-glucosidase from Botrytis cinerea. Enzyme Microb. Technol. 1995, 17, 900–906. [Google Scholar] [CrossRef]
- Teugjas, H.; Väljamäe, P. Selecting β-glucosidases to support cellulases in cellulose saccharification. Biotechnol. Biofuels 2013, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Rajasree, K.P.; Mathew, G.M.; Pandey, A.; Sukumaran, R.K. Highly glucose tolerant β-glucosidase from Aspergillus unguis: NII 08123 for enhanced hydrolysis of biomass. J. Ind. Microbiol. Biotechnol. 2013, 40, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, X.; Yin, Q.; Fang, W.; Fang, Z.; Wang, X.; Zhang, X.; Xiao, Y. A mechanism of glucose tolerance and stimulation of GH1 β-glucosidases. Sci. Rep. 2015, 5, 17296. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pang, Q.; Zhao, L.; Fan, S.; Shi, H. Thermoanaerobacterium thermosaccharolyticum β-glucosidase: A glucose-tolerant enzyme with high specific activity for cellobiose. Biotechnol. Biofuels 2012, 5, 1–10. [Google Scholar] [CrossRef]
- Mariano, D.C.B.; Leite, C.; Santos, L.H.S.; Marins, L.F.; Machado, K.S.; Werhli, A.V.; Lima, L.H.F.; de Melo-Minardi, R.C. Characterization of glucose-tolerant β-glucosidases used in biofuel production under the bioinformatics perspective: A systematic review. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef]
- Salgado, J.C.S.; Meleiro, L.P.; Carli, S.; Ward, R.J. Glucose tolerant and glucose stimulated β-glucosidases—A review. Bioresour. Technol. 2018, 267, 704–713. [Google Scholar] [CrossRef]
- Berrin, J.-G.; Czjzek, M.; Kroon, P.A.; McLauchlan, W.R.; Puigserver, A.; Williamson, G.; Juge, N. Substrate (aglycone) specificity of human cytosolic beta-glucosidase. Biochem. J. 2003, 373, 41–48. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Fang, Z.; Fang, W.; Peng, H.; Xiao, Y. The 184th residue of β-glucosidase Bgl1B plays an important role in glucose tolerance. J. Biosci. Bioeng. 2011, 112, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-L.; Chang, C.-K.; Jeng, W.-Y.; Wang, A.H.-J.; Liang, P.-H. Mutations in the substrate entrance region of -glucosidase from Trichoderma reesei improve enzyme activity and thermostability. Protein Eng. Des. Sel. 2012, 25, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Amano, Y.; Nozaki, K. Improvements in Glucose Sensitivity and Stability of Trichoderma reesei β-Glucosidase Using Site-Directed Mutagenesis. PLoS ONE 2016, 11, e0147301. [Google Scholar] [CrossRef] [PubMed]
- Sansenya, S.; Maneesan, J.; Cairns, J.R.K. Exchanging a single amino acid residue generates or weakens a +2 cellooligosaccharide binding subsite in rice β-glucosidases. Carbohydr. Res. 2012, 351, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Chuenchor, W.; Pengthaisong, S.; Robinson, R.C.; Yuvaniyama, J.; Oonanant, W.; Bevan, D.R.; Esen, A.; Chen, C.-J.; Opassiri, R.; Svasti, J.; et al. Structural insights into rice BGlu1 beta-glucosidase oligosaccharide hydrolysis and transglycosylation. J. Mol. Biol. 2008, 377, 1200–1215. [Google Scholar] [CrossRef] [PubMed]
- Zouhar, J.; Vévodová, J.; Marek, J.; Damborský, J.; Su, X.D.; Brzobohatý, B. Insights into the functional architecture of the catalytic center of a maize beta-glucosidase Zm-p60.1. Plant Physiol. 2001, 127, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Sansenya, S.; Opassiri, R.; Kuaprasert, B.; Chen, C.-J.; Cairns, J.R.K. The crystal structure of rice (Oryza sativa L.) Os4BGlu12, an oligosaccharide and tuberonic acid glucoside-hydrolyzing β-glucosidase with significant thioglucohydrolase activity. Arch. Biochem. Biophys. 2011, 510, 62–72. [Google Scholar] [CrossRef]
- Tsukada, T.; Igarashi, K.; Fushinobu, S.; Samejima, M. Role of subsite +1 residues in pH dependence and catalytic activity of the glycoside hydrolase family 1 beta-glucosidase BGL1A from the basidiomycete Phanerochaete chrysosporium. Biotechnol. Bioeng. 2008, 99, 1295–1302. [Google Scholar] [CrossRef]
- Sanz-Aparicio, J.; Hermoso, J.A.; Martínez-Ripoll, M.; Lequerica, J.L.; Polaina, J. Crystal structure of beta-glucosidase A from Bacillus polymyxa: Insights into the catalytic activity in family 1 glycosyl hydrolases. J. Mol. Biol. 1998, 275, 491–502. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Jo, T.; Uchiyama, T.; Manninen, J.A.; Arakawa, T.; Miyazaki, K.; Fushinobu, S.; Yaoi, K. Crystal structure and identification of a key amino acid for glucose tolerance, substrate specificity, and transglycosylation activity of metagenomic β-glucosidase Td2F2. FEBS J. 2016, 283, 2340–2353. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. In Data Mining: Practical Machine Learning Tools and Techniques; Morgan Kaufmann: Burlington, MA, USA, 2016. [Google Scholar]
- Silva, M.F.M.; Leijoto, L.F.; Nobre, C.N. Algorithms Analysis in Adjusting the SVM Parameters: An Approach in the Prediction of Protein Function. Appl. Artif. Intell. 2017, 31, 1–16. [Google Scholar] [CrossRef]
- Fang, W.; Fang, Z.; Liu, J.; Hong, Y.; Peng, H.; Zhang, X.; Sun, B.; Xiao, Y. Cloning and characterization of a beta-glucosidase from marine metagenome. Sheng Wu Gong Cheng Xue Bao 2009, 25, 1914–1920. [Google Scholar] [PubMed]
- Fang, Z.; Fang, W.; Liu, J.; Hong, Y.; Peng, H.; Zhang, X.; Sun, B.; Xiao, Y. Cloning and Characterization of a β-Glucosidase from Marine Microbial Metagenome with Excellent Glucose Tolerance. J. Microbiol. Biotechnol. 2010, 20, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, V.; Ebert, C.; Svendsen, A.; Besenmatter, W.; Gardossi, L. An integrated platform for automatic design and screening of virtual mutants based on 3D-QSAR analysis. J. Mol. Catal. B Enzym. 2014, 101, 7–15. [Google Scholar] [CrossRef]
- Braiuca, P.; Boscarol, L.; Ebert, C.; Linda, P.; Gardossi, L. 3D-QSAR Applied to the Quantitative Prediction of Penicillin G Amidase Selectivity. Adv. Synth. Catal. 2006, 348, 773–780. [Google Scholar] [CrossRef]
- Steinkellner, G.; Gruber, C.C.; Pavkov-Keller, T.; Binter, A.; Steiner, K.; Winkler, C.; Łyskowski, A.; Schwamberger, O.; Oberer, M.; Schwab, H.; et al. Identification of promiscuous ene-reductase activity by mining structural databases using active site constellations. Nat. Commun. 2014, 5, 4150. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- de Giuseppe, P.O.; Souza, T.d.A.C.B.; Souza, F.H.M.; Zanphorlin, L.M.; Machado, C.B.; Ward, R.J.; Jorge, J.A.; Furriel, R.d.P.M.; Murakami, M.T. Structural basis for glucose tolerance in GH1 β-glucosidases. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1631–1639. [Google Scholar] [CrossRef]
- Jeng, W.-Y.; Wang, N.-C.; Lin, C.-T.; Chang, W.-J.; Liu, C.-I.; Wang, A.H.-J. High-resolution structures of Neotermes koshunensis β-glucosidase mutants provide insights into the catalytic mechanism and the synthesis of glucoconjugates. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 829–838. [Google Scholar] [CrossRef]
- Shatsky, M.; Nussinov, R.; Wolfson, H.J. A method for simultaneous alignment of multiple protein structures. Proteins 2004, 56, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bitar, M.; Franco, G.R. A basic protein comparative three-dimensional modeling methodological workflow theory and practice. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2014, 47. [Google Scholar] [CrossRef] [PubMed]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Mutation | Effect | Classification | Source |
|---|---|---|---|---|
| 1 | H228T | Improves glucose tolerance. | Beneficial | [27] |
| 2 | V174C/A404V/L441F | Increases the optimal temperature of 50 °C to 60 °C, reduces the optimal pH of 6 to 5.5. | Beneficial | [3] |
| 3 | H184F | Increases the inhibition constant for glucose. | Beneficial | [32] |
| 4 | P172L | Increases catalytic efficiency. | Beneficial | [33] |
| 5 | P172L/F250A | Increases catalytic efficiency. | Beneficial | [33] |
| 6 | L167W | Increases the optimal temperature and glucose tolerance. | Beneficial | [33] |
| 7 | L167W/P172L | Increases the activity (2×). | Beneficial | [34] |
| 8 | L167W/P172L/P338F | Increases the activity (1,3×). | Beneficial | [34] |
| 9 | V168Y | Reduction in the specific activity. | Not beneficial | [31] |
| 10 | F225S | Reduction in the specific activity. | Not beneficial | [31] |
| 11 | Y308F | Reduction in the specific activity. | Not beneficial | [31] |
| 12 | Y308A | Reduction in the specific activity. | Not beneficial | [31] |
| 13 | I207V | Increases the specificity constant (Kcat/Km). | Beneficial | [35] |
| 14 | N218H | Decreases the Km about 2-fold. | Beneficial | [36] |
| 15 | N273V | Increases the Km about 5-fold. | Not beneficial | [36] |
| 16 | F252I | Reduces substrate affinity. | Not beneficial | [37] |
| 17 | F252W | Reduces substrate affinity. | Not beneficial | [37] |
| 18 | F252Y | Reduces substrate affinity. | Not beneficial | [37] |
| 19 | M284N | Reduction of Kcat/Km 7 to 30-fold depending on the substrate. | Not beneficial | [35] |
| 20 | H276M | Reduction of Kcat/Km 2 to 6-fold depending on the substrate. | Not beneficial | [38] |
| 21 | V173C | Decreases affinity for cellobiose. | Not beneficial | [39] |
| 22 | M177L | Decreases affinity for cellobiose (small reduction). | Not beneficial | [39] |
| 23 | D229N | Decreases affinity for cellobiose (high reduction). | Not beneficial | [39] |
| 24 | H231D | Decreases affinity for cellobiose. | Not beneficial | [39] |
| 25 | E96K | Improves the thermostability. | Beneficial | [40] |
| 26 | N223G | Reduction of transglycosylation, glucose tolerance, and activity. | Not beneficial | [41] |
| 27 | N223Q | Reduction of transglycosylation, glucose tolerance, and activity. | Not beneficial | [41] |
| ID | Mutation | ΔΔSSV Expected | ΔΔSSV Score | Hit |
|---|---|---|---|---|
| 1 | H228T | ΔΔSSV < 0 | −186.18 | ✓ |
| 2 | V174C/A404V/L441F | ΔΔSSV < 0 | −246.22 | ✓ |
| 3 | H184F | ΔΔSSV < 0 | 100.37 | |
| 4 | P172L | ΔΔSSV < 0 | −6.29 | ✓ |
| 5 | P172L/F250A | ΔΔSSV < 0 | −6.29 | ✓ |
| 6 | L167W | ΔΔSSV < 0 | −602.80 | ✓ |
| 7 | L167W/P172L | ΔΔSSV < 0 | −615.46 | ✓ |
| 8 | L167W/P172L/P338F | ΔΔSSV < 0 | −615.46 | ✓ |
| 9 | V168Y | ΔΔSSV > 0 | 330.56 | ✓ |
| 10 | F225S | ΔΔSSV > 0 | −365.07 | |
| 11 | Y308F | ΔΔSSV > 0 | 34.19 | ✓ |
| 12 | Y308A | ΔΔSSV > 0 | −108.62 | |
| 13 | I207V | ΔΔSSV < 0 | −71.56 | ✓ |
| 14 | N218H | ΔΔSSV < 0 | −230.61 | ✓ |
| 15 | N273V | ΔΔSSV > 0 | −55.26 | |
| 16 | F252I | ΔΔSSV > 0 | 86.70 | ✓ |
| 17 | F252W | ΔΔSSV > 0 | 129.97 | ✓ |
| 18 | F252Y | ΔΔSSV > 0 | 37.86 | ✓ |
| 19 | M284N | ΔΔSSV > 0 | −127.35 | |
| 20 | H276M | ΔΔSSV > 0 | −501.32 | |
| 21 | V173C | ΔΔSSV > 0 | 13.59 | ✓ |
| 22 | M177L | ΔΔSSV > 0 | 20.86 | ✓ |
| 23 | D229N | ΔΔSSV > 0 | 18.11 | ✓ |
| 24 | H231D | ΔΔSSV > 0 | −54.22 | |
| 25 | E96K | ΔΔSSV < 0 | −31.08 | ✓ |
| 26 | N223G | ΔΔSSV > 0 | 39.37 | ✓ |
| 27 | N223Q | ΔΔSSV > 0 | 264.34 | ✓ |
| Metric | SSV | SVM (Wild) | SVM (Mutant) | SVM (Wild-Mutant) |
|---|---|---|---|---|
| Precision | 0.89 | 0.64 | 0.36 | 0.36 |
| Accuracy | 0.74 | 0.81 | 0.74 | 0.74 |
| Specificity | 0.92 | 0.79 | 0.70 | 0.70 |
| Sensitivity | 0.57 | 0.88 | 1.00 | 1.00 |
| F-measure | 0.70 | 0.74 | 0.53 | 0.53 |
| F172 | G246 | H228 | T299 | V227 |
|---|---|---|---|---|
| F172I | G246S | H228A | T299S | V227M |
| F172K | G246T | H228C | ||
| F172V | H228M | |||
| H228N | ||||
| H228P | ||||
| H228Q | ||||
| H228T | ||||
| H228V |
| Mutant | Mutation | IF | Classification | ΔΔSSV | Hit |
|---|---|---|---|---|---|
| M1 | G39A/W104F/L278A | 6.3 | Beneficial | −841 | ✓ |
| M2 | G39A/T103G/L278A | 3.8 | Beneficial | −121 | ✓ |
| M3 | G39A/T103G/W104F/L278A | 11.2 | Beneficial | −841 | ✓ |
| M4 | G39A | 2.8 | Beneficial | 150 | |
| M5 | G39A/L278A | 3.3 | Beneficial | −121 | ✓ |
| M6 | I189A | 0.4 | Not beneficial | −94 | |
| M7 | T40A | 0.4 | Not beneficial | 40 | ✓ |
| M8 | T103G | 1.1 | Neutral/Beneficial | 0 | ✓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mariano, D.C.B.; Santos, L.H.; Machado, K.d.S.; Werhli, A.V.; de Lima, L.H.F.; de Melo-Minardi, R.C. A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV). Int. J. Mol. Sci. 2019, 20, 333. https://doi.org/10.3390/ijms20020333
Mariano DCB, Santos LH, Machado KdS, Werhli AV, de Lima LHF, de Melo-Minardi RC. A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV). International Journal of Molecular Sciences. 2019; 20(2):333. https://doi.org/10.3390/ijms20020333
Chicago/Turabian StyleMariano, Diego César Batista, Lucianna Helene Santos, Karina dos Santos Machado, Adriano Velasque Werhli, Leonardo Henrique França de Lima, and Raquel Cardoso de Melo-Minardi. 2019. "A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV)" International Journal of Molecular Sciences 20, no. 2: 333. https://doi.org/10.3390/ijms20020333
APA StyleMariano, D. C. B., Santos, L. H., Machado, K. d. S., Werhli, A. V., de Lima, L. H. F., & de Melo-Minardi, R. C. (2019). A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV). International Journal of Molecular Sciences, 20(2), 333. https://doi.org/10.3390/ijms20020333