Etiology of Metabolic Syndrome and Dietary Intervention

Abstract
1. Introduction
2. Etiology of MetS
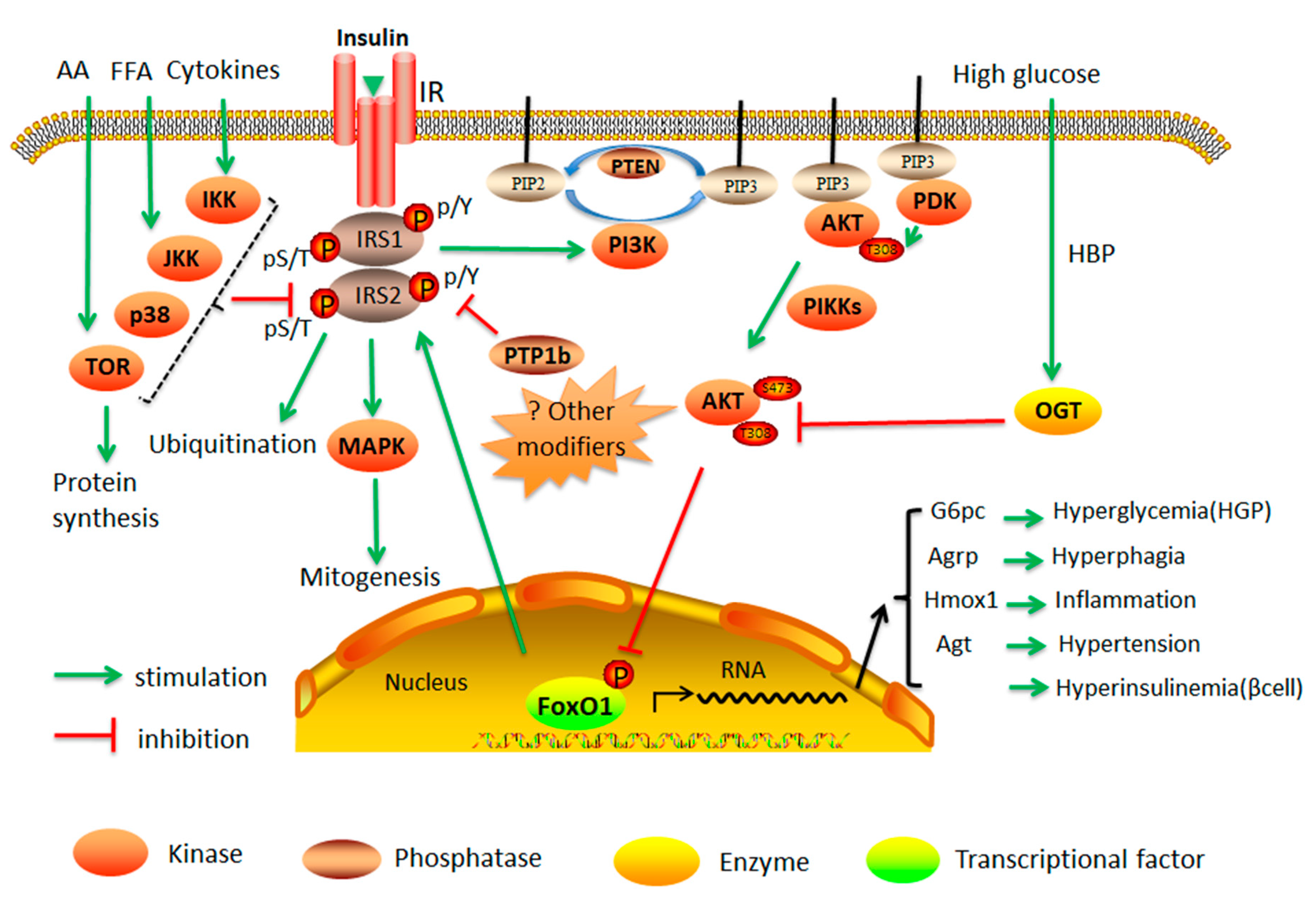
2.1. Insulin Resistance
2.2. Pancreatic β-Cell Dysfunction
2.3. Cellular Dysfunction by Protein Kinases and Phosphatases
2.4. Suppression of IRS1 and IRS2 Gene Expression and Function
2.5. Obesity and Lipid Toxicity
2.6. Oxidative Stress and Glucose Toxicity
2.7. Chronic Inflammation
2.8. Circadian
2.9. Genetics and Epigenetics
2.10. Gut Microbiota
2.11. Dietary Effects
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reaven, G.M. Role of Insulin Resistance in Human Disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D.; Karnieli, E. The metabolic syndrome—From insulin resistance to obesity and diabetes. Endocrinol. Metab. Clin. N. Am. 2008, 37, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Reaven, G.M. The metabolic syndrome: Is this diagnosis necessary ? Am. J. Clin. Nutr. 2006, 83, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Fryar, C.D.; Flegal, K.M. Prevalence of Obesity Among Adults and Youth: United States, 2011–2014. NCHS Data Brief 2015, 219, 1–8. [Google Scholar]
- Obesity and Overweight, Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 1 August 2018).
- Ervin, R.B. Prevalence of Metabolic Syndrome among Adults 20 Years of Age and Over, by Sex, Age, Race and Ethnicity, and Body Mass Index: United States, 2003–2006; National Health Statistics Reports; U.S. Department of Health and Human Services: Washington, DC, USA, 2009; pp. 1–7.
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the Metabolic Syndrome in the United States, 2003–2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Soni, K.G.; Semache, M.; Casavant, S.; Fortier, M.; Pan, L.; Mitchell, G.A. Lipolysis and the integrated physiology of lipid energy metabolism. Mol. Genet. Metab. 2008, 95, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, D.; Reaven, G.M.; Cobin, R.H.; Ford, E.; Ganda, O.P.; Handelsman, Y.; Hellman, R.; Jellinger, P.S.; Kendall, D.; Krauss, R.M. American College of Endocrinology Position Statement on the Insulin Resistance Syndrome. Endocr. Pract. 2003, 9, 237–252. [Google Scholar] [PubMed]
- Albert, V.; Svensson, K.; Shimobayashi, M.; Colombi, M.; Munoz, S.; Jimenez, V.; Handschin, C.; Bosch, F.; Hall, M.N. mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol. Med. 2016, 8, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Zimmet, P.; Shaw, J. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Guo, S.D. Insulin signaling, resistance, and metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef] [PubMed]
- Karnieli, E.; Armoni, M. Transcriptional regulation of the insulin-responsive glucose transporter GLUT4 gene: From physiology to pathology. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E38–E45. [Google Scholar] [CrossRef] [PubMed]
- Armoni, M.; Harel, C.; Karnieli, E. Transcriptional regulation of the GLUT4 gene: From PPAR-gamma and FOXO1 to FFA and inflammation. Trends Endocrinol. Metab. 2007, 18, 100–107. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D. Beta-cell dysfunction and insulin resistance in type 2 diabetes—Role of metabolic and genetic abnormalities. Am. J. Med. 2002, 113, 3S–11S. [Google Scholar] [CrossRef]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 2001, 50, S154–S159. [Google Scholar] [CrossRef] [PubMed]
- Malin, S.K.; Finnegan, S.; Fealy, C.E.; Filion, J.; Rocco, M.B.; Kirwan, J.P. beta-Cell dysfunction is associated with metabolic syndrome severity in adults. Metab. Syndr. Relat. Disord. 2014, 12, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.S.; Dalleck, L.C.; Borrani, F.; Fassett, R.G.; Coombes, J.S. Cardiorespiratory fitness is positively associated with increased pancreatic beta cell function independent of fatness in individuals with the metabolic syndrome: Fitness versus fatness. J. Sci. Med. Sport 2017, 20, 45–49. [Google Scholar] [CrossRef]
- Barry, V.W.; Baruth, M.; Beets, M.W.; Durstine, J.L.; Liu, J.; Blair, S.N. Fitness vs. fatness on all-cause mortality: A meta-analysis. Prog. Cardiovasc. Dis. 2014, 56, 382–390. [Google Scholar] [CrossRef]
- Lee, D.C.; Sui, X.; Artero, E.G.; Lee, I.M.; Church, T.S.; McAuley, P.A.; Stanford, F.C.; Kohl, H.W., 3rd; Blair, S.N. Long-term effects of changes in cardiorespiratory fitness and body mass index on all-cause and cardiovascular disease mortality in men: The Aerobics Center Longitudinal Study. Circulation 2011, 124, 2483–2490. [Google Scholar] [CrossRef]
- De Schutter, A.; Lavie, C.J.; Milani, R.V. The impact of obesity on risk factors and prevalence and prognosis of coronary heart disease-the obesity paradox. Prog. Cardiovasc. Dis. 2014, 56, 401–408. [Google Scholar] [CrossRef]
- Han, S.S.; Kim, K.W.; Kim, K.I.; Na, K.Y.; Chae, D.W.; Kim, S.; Chin, H.J. Lean mass index: A better predictor of mortality than body mass index in elderly Asians. J. Am. Geriatr. Soc. 2010, 58, 312–317. [Google Scholar] [CrossRef]
- Srikanthan, P.; Karlamangla, A.S. Muscle mass index as a predictor of longevity in older adults. Am. J. Med. 2014, 127, 547–553. [Google Scholar] [CrossRef]
- Lee, C.G.; Boyko, E.J.; Nielson, C.M.; Stefanick, M.L.; Bauer, D.C.; Hoffman, A.R.; Dam, T.T.; Lapidus, J.A.; Cawthon, P.M.; Ensrud, K.E.; et al. Mortality risk in older men associated with changes in weight, lean mass, and fat mass. J. Am. Geriatr. Soc. 2011, 59, 233–240. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Xue, G.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar] [PubMed]
- Guo, C.A.; Guo, S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J. Endocrinol. 2017, 233, R131–R143. [Google Scholar] [CrossRef] [PubMed]
- Furtado, L.M.; Somwar, R.; Sweeney, G.; Niu, W.; Klip, A. Activation of the Glucose Transporter GLUT4 by Insulin. Biochem. Cell Biol. 2002, 80, 569–578. [Google Scholar] [CrossRef]
- Michelle Furtado, L.; Poon, V.; Klip, A. GLUT4 activation: Thoughts on possible mechanisms. Acta Physiol. Scand. 2003, 178, 287–296. [Google Scholar] [CrossRef]
- Houde, V.P.; Brûlé, S.; Festuccia, W.T.; Blanchard, P.-G.; Bellmann, K.; Deshaies, Y.; Marette, A. Chronic Rapamycin Treatment Causes Glucose Intolerance and Hyperlipidemia by Upregulating Hepatic Gluconeogenesis and Impairing Lipid Deposition in Adipose Tissue. Diabetes 2010, 59, 1338–1348. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharm. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yu, Y.; Zhang, Z.; Chen, X.; Hu, Z.; Tong, Q.; Chang, J.; Feng, X.H.; Lin, X. SCP4 Promotes Gluconeogenesis through FoxO1/3a Dephosphorylation. Diabetes 2017. [Google Scholar] [CrossRef] [PubMed]
- Withers, D.J.; Burks, D.J.; Towery, H.H.; Altamuro, S.L.; Flint, C.L.; White, M.F. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat. Genet. 1999, 23, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M.; et al. Responses of Gut Microbiota and Glucose and Lipid Metabolism to Prebiotics in Genetic Obese and Diet-Induced Leptin-Resistant Mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M. Insulin resistance and hypertension: New insights. Kidney Int. 2015, 87, 497–499. [Google Scholar] [CrossRef]
- Ryu, J.; Galan, A.K.; Xin, X.; Dong, F.; Abdul-Ghani, M.A.; Zhou, L.; Wang, C.; Li, C.; Holmes, B.M.; Sloane, L.B.; et al. APPL1 Potentiates Insulin Sensitivity by Facilitating the Binding of IRS1/2 to the Insulin Receptor. Cell Rep. 2014, 7, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, J.; Chandarlapaty, S.; Cross, J.; Thompson, C.; Rosen, N.; Jiang, X. PTEN is a protein tyrosine phosphatase for IRS1. Nat. Struct. Mol. Biol. 2014, 21, 522–527. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Nanhwan, M.K.; Ling, S.; Perez-Polo, J.R.; Ye, Y.; Bajaj, M. PTEN upregulation may explain the development of insulin resistance and type 2 diabetes with high dose statins. Cardiovasc. Drugs 2014, 28, 447–457. [Google Scholar] [CrossRef]
- Hakuno, F.; Fukushima, T.; Yoneyama, Y.; Kamei, H.; Ozoe, A.; Yoshihara, H.; Yamanaka, D.; Shibano, T.; Sone-Yonezawa, M.; Yu, B.C.; et al. The Novel Functions of High-Molecular-Mass Complexes Containing Insulin Receptor Substrates in Mediation and Modulation of Insulin-Like Activities: Emerging Concept of Diverse Functions by IRS-Associated Proteins. Front. Endocrinol. 2015, 6, 73. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Chen, J. MEMO1, a new IRS1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells. Oncogene 2013, 32, 3130–3138. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Okajima, H.; Yamanaka, D.; Ariga, M.; Nagata, S.; Ito, A.; Yoshida, M.; Asano, T.; Chida, K.; Hakuno, F.; et al. HSP90 interacting with IRS-2 is involved in cAMP-dependent potentiation of IGF-I signals in FRTL-5 cells. Mol. Cell. Endocrinol. 2011, 344, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Yoshihara, H.; Furuta, H.; Kamei, H.; Hakuno, F.; Luan, J.; Duan, C.; Saeki, Y.; Tanaka, K.; Iemura, S.; et al. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat. Commun. 2015, 6, 6780. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Tobe, K.; Terauchi, Y.; Eto, K.; Yamauchi, T.; Suzuki, R.; Tsubamoto, Y.; Komeda, K.; Nakano, R.; Miki, H.; et al. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 2000, 49, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Withers, D.J.; Gutierrez, J.S.; Towery, H.; Burks, D.J.; Ren, J.; Previs, S.; Zhang, Y.; Bernal, D.; Pons, S.; Shulman, G.; et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998, 391, 900–904. [Google Scholar] [CrossRef]
- Araki, E.; Lipes, M.A.; Patti, M.-E.; Brüning, J.C.; Haag III, B.; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.R.; Chen, Y.; McMullen, M.F.; et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef]
- Parvaiz, F.; Manzoor, S.; Iqbal, J.; Sarkar-Dutta, M.; Imran, M.; Waris, G. Hepatitis C virus NS5A promotes insulin resistance through IRS-1 serine phosphorylation and increased gluconeogenesis. World J. Gastroenterol. 2015, 21, 12361–12369. [Google Scholar] [CrossRef]
- Sayaka Katagiri, K.P.; Maeda, Y.; Rao, T.N.; Khamaisi, M.; Li, Q.; Yokomizo, H.; Mima, A.; Lancerotto, L.; Wagers, A.; Orgill, D.P.; et al. Overexpressing IRS1 in Endothelial Cells Enhances Angioblast Differentiation and Wound Healing in Diabetes and Insulin Resistance. Diabetes 2016, 65, 2760–2771. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Motohashi, N.; Alexander, M.S.; Shimizu-Motohashi, Y.; Myers, J.A.; Kawahara, G.; Kunkel, L.M. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. J. Cell Sci. 2013, 126, 2678–2691. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, C.; Cheng, J.; Chen, B.; Ke, Q.; Lv, Z.; Wu, J.; Zhou, Y. MicroRNA-145 suppresses hepatocellular carcinoma by targeting IRS1 and its downstream Akt signaling. Biochem. Biophys. Res. Commun. 2014, 446, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Alfaradhi, M.Z.; Martin-Gronert, M.S.; Duque-Guimaraes, D.E.; Piekarz, A.; Ferland-McCollough, D.; Bushell, M.; Ozanne, S.E. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol. Metab. 2014, 3, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Tobe, K.; Yano, W.; Suzuki, R.; Ueki, K.; Takamoto, I.; Satoh, H.; Maki, T.; Kubota, T.; et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J. Clin. Investig. 2004, 114, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Kubota, N.; Sato, H.; Sasaki, M.; Takamoto, I.; Kubota, T.; Nakaya, K.; Noda, M.; Ueki, K.; Kadowaki, T. Insulin receptor substrate-2 (Irs2) in endothelial cells plays a crucial role in insulin secretion. Diabetes 2015, 64, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Li, Q.; Rask-Madsen, C.; Mima, A.; Mizutani, K.; Winnay, J.; Maeda, Y.; D’Aquino, K.; White, M.F.; Feener, E.P.; et al. Serine phosphorylation sites on IRS2 activated by angiotensin II and protein kinase C to induce selective insulin resistance in endothelial cells. Mol. Cell. Biol. 2013, 33, 3227–3241. [Google Scholar] [CrossRef]
- Neukamm, S.S.; Ott, J.; Dammeier, S.; Lehmann, R.; Haring, H.U.; Schleicher, E.; Weigert, C. Phosphorylation of serine 1137/1138 of mouse insulin receptor substrate (IRS) 2 regulates cAMP-dependent binding to 14-3-3 proteins and IRS2 protein degradation. J. Biol. Chem. 2013, 288, 16403–16415. [Google Scholar] [CrossRef]
- Day, E.; Poulogiannis, G.; McCaughan, F.; Mulholland, S.; Arends, M.J.; Ibrahim, A.E.; Dear, P.H. IRS2 is a candidate driver oncogene on 13q34 in colorectal cancer. Int. J. Exp. Pathol. 2013, 94, 203–211. [Google Scholar]
- Guo, S. Molecular Basis of Insulin Resistance: The Role of IRS and Foxo1 in the Control of Diabetes Mellitus and Its Complications. Drug Discov. Today Dis. Mech. 2013, 10, e27–e33. [Google Scholar] [CrossRef]
- Allison, M.B.; Myers, M.G., Jr. 20 years of leptin: Connecting leptin signaling to biological function. J. Endocrinol. 2014, 223, T25–T35. [Google Scholar] [CrossRef]
- Morris, D.L.; Rui, L.Y. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of Leptin Action and Leptin Resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Yiannikouris, F.; Karounos, M.; Charnigo, R.; English, V.L.; Rateri, D.L.; Daugherty, A.; Cassis, L.A. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R244–R251. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Guo, Z.; Randall, D.C.; Cassis, L.; Brown, D.R.; Gong, M.C. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1634–H1641. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, K.; Wu, Y.; Xu, Z.; Yong, Q.C.; Kumar, R.; Baker, K.M.; Zhu, Q.; Chen, S.; Guo, S. Novel mechanism of blood pressure regulation by forkhead box class O1-mediated transcriptional control of hepatic angiotensinogen. Hypertension 2014, 64, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.-H.; et al. Akt Stimulates Hepatic SREBP1c and Lipogenesis through Parallel mTORC1-Dependent and Independent Pathways. Cell Metab. 2011, 14, 280. [Google Scholar] [CrossRef]
- Jiang, F.; Lim, H.K.; Morris, M.J.; Prior, L.; Velkoska, E.; Wu, X.; Dusting, G.J. Systemic upregulation of NADPH oxidase in diet-induced obesity in rats. Redox Rep. 2011, 16, 223–229. [Google Scholar] [CrossRef]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of Obesity and Metabolic Syndrome on Immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.A.; Devaraj, S.; Afify, A.; Jialal, I. Adipose tissue dysregulation in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E1782–E1788. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, H.; Woo, S.L.; Kim, S.M.; Shende, V.R.; Neuendorff, N.; Guo, X.; Guo, T.; Qi, T.; Pei, Y.; et al. Myeloid cell-specific disruption of Period1 and Period2 exacerbates diet-induced inflammation and insulin resistance. J. Biol. Chem. 2014, 289, 16374–16388. [Google Scholar] [CrossRef]
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.T.; Depinho, R.A.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 2014, 158, 25–40. [Google Scholar] [CrossRef]
- Jialal, I.; Kaur, H.; Devaraj, S. Toll-like receptor status in obesity and metabolic syndrome: A translational perspective. J. Clin. Endocrinol. Metab. 2014, 99, 39–48. [Google Scholar] [CrossRef]
- Catrysse, L.; van Loo, G. Inflammation and the Metabolic Syndrome: The Tissue-Specific Functions of NF-kappaB. Trends Cell Biol. 2017, 27, 417–429. [Google Scholar] [CrossRef]
- Botchlett, R.; Woo, S.L.; Liu, M.; Pei, Y.; Guo, X.; Li, H.; Wu, C. Nutritional approaches for managing obesity-associated metabolic diseases. J. Endocrinol. 2017, 233, R145–R171. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 Fatty Acids and Inflammatory Processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Itariu, B.K.; Zeyda, M.; Hochbrugger, E.E.; Neuhofer, A.; Prager, G.; Schindler, K.; Bohdjalian, A.; Mascher, D.; Vangala, S.; Schranz, M.; et al. Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 96, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.; Finlin, B.S.; Unal, R.; Zhu, B.; Morris, A.J.; Shipp, L.R.; Lee, J.; Walton, R.G.; Adu, A.; Erfani, R.; et al. Kern. Omega-3 Fatty Acids Reduce Adipose Tissue Macrophages in Human Subjects With Insulin Resistance. Diabetes 2013, 62, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Li, F.; Liu, S.; Jin, Y.; Zhang, X.; Yang, T.; Dai, Y.; Li, X.; Zhao, A.Z. omega-3 polyunsaturated fatty acids ameliorate type 1 diabetes and autoimmunity. J. Clin. Investig. 2017, 127, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Garaulet, M.; Madrid, J.A. Chronobiological aspects of nutrition, metabolic syndrome and obesity. Adv. Drug Deliv. Rev. 2010, 62, 967–978. [Google Scholar] [CrossRef]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef]
- Chaves, I.; van der Horst, G.T.J.; Schellevis, R.; Nijman, R.M.; Koerkamp, M.G.; Holstege, F.C.P.; Smidt, M.P.; Hoekman, M.F.M. Insulin-FOXO3 Signaling Modulates Circadian Rhythms via Regulation of Clock Transcription. Curr. Biol. 2014, 24, 1248–1255. [Google Scholar] [CrossRef]
- Rudic, R.D.; McNamara, P.; Curtis, A.M.; Boston, R.C.; Panda, S.; Hogenesch, J.B.; FitzGerald, G.A. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004, 2, 1893–1899. [Google Scholar] [CrossRef]
- Shi, S.-Q.; Ansari, T.S.; McGuinness, O.P.; Wasserman, D.H.; Johnson, C.H. Circadian Disruption Leads to Insulin Resistance and Obesity. Curr. Biol. 2013, 23, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Hsu, J.W.; Manka, P.P.; Syn, W.K. Role of the Circadian Clock in the Metabolic Syndrome and Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2018, 63, 3187–3206. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; la Fleur, S.; Fliers, E. Circadian control of glucose metabolism. Mol. Metab. 2014, 3, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S.; Hong, H.-K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.L.; Patel, V.R.; de Mateo, S.; Orozco-Solis, R.; Ceglia, N.J.; Sahar, S.; Dilag-Penilla, S.A.; Dyar, K.A.; Baldi, P.; Sassone-Corsi, P. Reprogramming of the Circadian Clock by Nutritional Challenge. Cell 2013, 155, 1464–1478. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Pevet, P.; Challet, E. High-fat feeding alters the clock synchronization to light. J. Physiol. 2008, 586, 5901–5910. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-Fat Diet Disrupts Behavioral and Molecular Circadian Rhythms in Mice. Cell Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef]
- Young, M.E.; Wilson, C.R.; Razeghi, P.; Guthrie, P.H.; Taegtmeyer, H. Alterations of the circadian clock in the heart by streptozotocin-induced diabetes. J. Mol. Cell. Cardiol. 2002, 34, 223–231. [Google Scholar] [CrossRef]
- Fumagalli, M.; Moltke, I.; Grarup, N.; Racimo, F.; Bjerregaard, P.; Jorgensen, M.E.; Korneliussen, T.S.; Gerbault, P.; Skotte, L.; Linneberg, A.; et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 2015, 349, 1343–1347. [Google Scholar] [CrossRef]
- Zheng, J.S.; Parnell, L.D.; Smith, C.E.; Lee, Y.C.; Jamal-Allial, A.; Ma, Y.; Li, D.; Tucker, K.L.; Ordovas, J.M.; Lai, C.Q. Circulating 25-hydroxyvitamin D, IRS1 variant rs2943641, and insulin resistance: Replication of a gene-nutrient interaction in 4 populations of different ancestries. Clin. Chem. 2014, 60, 186–196. [Google Scholar] [CrossRef]
- Guo, S.; Cichy, S.B.; He, X.; Yang, Q.; Ragland, M.; Ghosh, A.K.; Johnson, P.F.; Unterman, T.G. Insulin suppresses transactivation by CAAT/enhancer-binding proteins beta (C/EBPbeta). Signaling to p300/CREB-binding protein by protein kinase B disrupts interaction with the major activation domain of C/EBPbeta. J. Biol. Chem. 2001, 276, 8516–8523. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Peng, J.; An, H.; He, Q.; Boronina, T.; Guo, S.; White, M.F.; Cole, P.A.; He, L. Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat. Commun. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Remely, M.; Haslberger, A.G. The microbial epigenome in metabolic syndrome. Mol. Asp. Med. 2017, 54, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Miles-Brown, J.; Pellizzon, M.; Ulman, E.; Ricci, M.; Zhang, L.; Patterson, A.D.; Vijay-Kumar, M.; Gewirtz, A.T. Lack of soluble fiber drives diet-induced adiposity in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G528–G541. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Potu, R.; Lu, H.; Vezzoni de Almeida, V.; Stewart, T.; Ragland, D.; Armstrong, A.; Adeola, O.; Nakatsu, C.H.; Ajuwon, K.M. Dietary fat content and fiber type modulate hind gut microbial community and metabolic markers in the pig. PLoS ONE 2013, 8, e59581. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; van-Hylckama Vlieg, J.E.; Strissel, K.; Zhao, L.; Obin, M.; et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J 2015, 9, 1–15. [Google Scholar] [CrossRef]
- Hulston, C.J.; Churnside, A.A.; Venables, M.C. Probiotic supplementation prevents high-fat, overfeeding-induced insulin resistance in human subjects. Br. J. Nutr. 2015, 113, 596–602. [Google Scholar] [CrossRef]
- Lye, H.S.; Kuan, C.Y.; Ewe, J.A.; Fung, W.Y.; Liong, M.T. The Improvement of Hypertension by Probiotics: Effects on Cholesterol, Diabetes, Renin, and Phytoestrogens. Int. J. Mol. Sci. 2009, 10, 3755–3775. [Google Scholar] [CrossRef]
- Guenther Boden, C.H.; Barrero, C.A.; Stein, T.P.; Chen, X.; Cheung, P.; Fecchio, C.; Koller, S.; Merali, S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci. Transl. Med. 2015, 7, 304re307. [Google Scholar]
- Raziani, F.; Tholstrup, T.; Kristensen, M.D.; Svanegaard, M.L.; Ritz, C.; Astrup, A.; Raben, A. High intake of regular-fat cheese compared with reduced-fat cheese does not affect LDL cholesterol or risk markers of the metabolic syndrome: A randomized controlled trial. Am. J. Clin. Nutr. 2016, 104, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, R.; Hostmark, A.T.; Haug, A.; Skeie, S. Effect of a high intake of cheese on cholesterol and metabolic syndrome: Results of a randomized trial. Food Nutr. Res. 2015, 59, 27651. [Google Scholar] [CrossRef] [PubMed]
- Thorning, T.K.; Raziani, F.; Bendsen, N.T.; Astrup, A.; Tholstrup, T.; Raben, A. Diets with high-fat cheese, high-fat meat, or carbohydrate on cardiovascular risk markers in overweight postmenopausal women: A randomized crossover trial. Am. J. Clin. Nutr. 2015, 102, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Villani, V.; Buono, R.; Wei, M.; Kumar, S.; Yilmaz, O.H.; Cohen, P.; Sneddon, J.B.; Perin, L.; Longo, V.D. Fasting-Mimicking Diet Promotes Ngn3-Driven beta-Cell Regeneration to Reverse Diabetes. Cell 2017, 168, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hruby, A.; Bernstein, A.M.; Ley, S.H.; Wang, D.D.; Chiuve, S.E.; Sampson, L.; Rexrode, K.M.; Rimm, E.B.; Willett, W.C.; et al. Saturated Fats Compared With Unsaturated Fats and Sources of Carbohydrates in Relation to Risk of Coronary Heart Disease: A Prospective Cohort Study. J. Am. Coll. Cardiol. 2015, 66, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated Fatty Acid–Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome–Mediated IL-1β Secretion and Insulin Resistance Despite Obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- Pinent, M.; Cedo, L.; Montagut, G.; Blay, M.; Ardevol, A. Procyanidins Improve some Disrupted Glucose Homoeostatic Situations: An Analysis of Doses and Treatments According to Different Animal Models. Crit. Rev. Food Sci. Nutr. 2012, 52, 569–584. [Google Scholar] [CrossRef]
- Babu, P.V.A.; Liu, D.M.; Gilbert, E.R. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J. Nutr. Biochem. 2013, 24, 1777–1789. [Google Scholar] [CrossRef]
- Li, X.; Sui, Y.; Li, S.; Xie, B.; Sun, Z. A-type procyanidins from litchi pericarp ameliorate hyperglycaemia by regulating hepatic and muscle glucose metabolism in streptozotocin (STZ)-induced diabetic mice fed with high fat diet. J. Funct. Foods 2016, 27, 711–722. [Google Scholar] [CrossRef]
- Li, X.; Li, S.; Chen, M.; Wang, J.; Xie, B.; Sun, Z. (-)-Epigallocatechin-3-Gallate (EGCG) inhibits starch digestion and improves glucose homeostasis through direct or indirect activation of PXR/CAR-mediated phase II metabolism in diabetic mice. Food Funct. 2018, 9, 4651–4663. [Google Scholar] [CrossRef] [PubMed]
- Blade, C.; Arola, L.; Salvado, M.J. Hypolipidemic effects of proanthocyanidins and their underlying biochemical and molecular mechanisms. Mol. Nutr. Food Res. 2010, 54, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Hanhineva, K.; Törrönen, R.; Bondia-Pons, I.; Pekkinen, J.; Kolehmainen, M.; Mykkänen, H.; Poutanen, K. Impact of Dietary Polyphenols on Carbohydrate Metabolism. Int. J. Mol. Sci. 2010, 11, 1365–1402. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, W.; Chen, H.; Liao, N.; Wang, Z.; Zhang, X.; Hai, C. High selenium impairs hepatic insulin sensitivity through opposite regulation of ROS. Toxicol. Lett. 2014, 224, 16–23. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Evaluation Content | WHO 1998 | EGIR 19 99 | NCEPATPIII 2001 | AACE 2003 | NCEP ATPIII 2005 | AHA/NHLBI 2005 | IDF 2005 | IDF/NHLBI 2009 |
|---|---|---|---|---|---|---|---|---|
| Criteria | IGT, IFG, T2D, or reduced insulin sensitivity plus any two of the following | Plasma insulin > 75 percentile plus any two of the following | Any three of the following | IGT or IFG plus any of the following | Any three of the following | Any three of the following | Increased WC plus any two of the following | Three out of five of the following |
| Obesity | Men: WHR > 0.9; Women: WHR > 0.85 and/or BMI > 30 kg/m2 | WC ≥ 94 cm in men or ≥ 80 cm in women | WC ≥ 102 cm in men or ≥ 88 cm in women | BMI > 25 kg/m2 | WC ≥ 102 cm in men or ≥ 88 cm in women | WC ≥ 102 cm in men or ≥ 88 cm in women | population-specific increased WC cutoffs | population-and country-specific WC cutoffs |
| Glucose | IGT, IGF, or T2D | IGT or IFG | ≥110 mg/dL (including T2D) | IGT or IFG | ≥100 mg/dL (including T2D) | ≥100 mg/dL or on drug treatment for elevated glucose | ≥100 mg/dL (including T2D) | ≥100 mg/dL |
| Triglycerides (TG) | TG ≥ 150 mg/dL | TG ≥ 150 mg/dL | TG ≥ 150 mg/dL | TG ≥ 150 mg/dL | TG ≥ 150 mg/dL or on therapy lowering TG | TG ≥ 150 mg/dL or on drug treatment for elevated triglycerides | TG ≥ 150 mg/dL or on therapy lowering TG | TG ≥ 150 mg/dL |
| High density lipoprotein (HDL)-cholesterol (HDL-C) | HDL-C < 40 mg/dLin men or HDL-C < 50 mg/dL in women | HDL-C < 39 mg/dL in men or women | HDL-C < 40 mg/dL in men or HDL-C < 50 mg/dL in women | HDL-C < 40 mg/dL in men or HDL-C < 50 mg/dL in women | HDL-C <40 mg/dL in men or HDL-C < 50 mg/dL in women on therapy increasing HDL-C | HDL-C < 40 mg/dL in men or HDL-C < 50 mg/dL in women or on drug treatment for reduced HDL-C | HDL-C < 40 mg/dL in men or HDL-C < 50 mg/dL in women on therapy increasing HDL-C | HDL-C< 40 mg/dL in men or HDL-C < 50 mg/dL in women |
| Blood pressure | ≥140/90 mmHg | ≥140/90 mmHg or on antihypertensive therapy | ≥130/85 mmHg | ≥130/85 mmHg | ≥130/85 mmHg or on antihypertensive therapy | ≥130/85 mmHg or on antihypertensive therapy | ≥130/85 mmHg or on antihypertensive therapy | ≥130/85 mmHg or on antihypertensive therapy |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.; Li, X.; Adams, H.; Kubena, K.; Guo, S. Etiology of Metabolic Syndrome and Dietary Intervention. Int. J. Mol. Sci. 2019, 20, 128. https://doi.org/10.3390/ijms20010128
Xu H, Li X, Adams H, Kubena K, Guo S. Etiology of Metabolic Syndrome and Dietary Intervention. International Journal of Molecular Sciences. 2019; 20(1):128. https://doi.org/10.3390/ijms20010128
Chicago/Turabian StyleXu, Hang, Xiaopeng Li, Hannah Adams, Karen Kubena, and Shaodong Guo. 2019. "Etiology of Metabolic Syndrome and Dietary Intervention" International Journal of Molecular Sciences 20, no. 1: 128. https://doi.org/10.3390/ijms20010128
APA StyleXu, H., Li, X., Adams, H., Kubena, K., & Guo, S. (2019). Etiology of Metabolic Syndrome and Dietary Intervention. International Journal of Molecular Sciences, 20(1), 128. https://doi.org/10.3390/ijms20010128